Mechanochemical Synthesis of Metallophenylsiloxanes Based on Polyphenylsiloxane and Acetylacetonates of Rare Earth Metals

Mechanochemical Synthesis of Metallophenylsiloxanes Based on Polyphenylsiloxane and Acetylacetonates of Rare Earth Metals

Received: 08 March 2026 Revised: 14 April 2026 Accepted: 01 June 2026 Published: 24 June 2026

© 2026 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
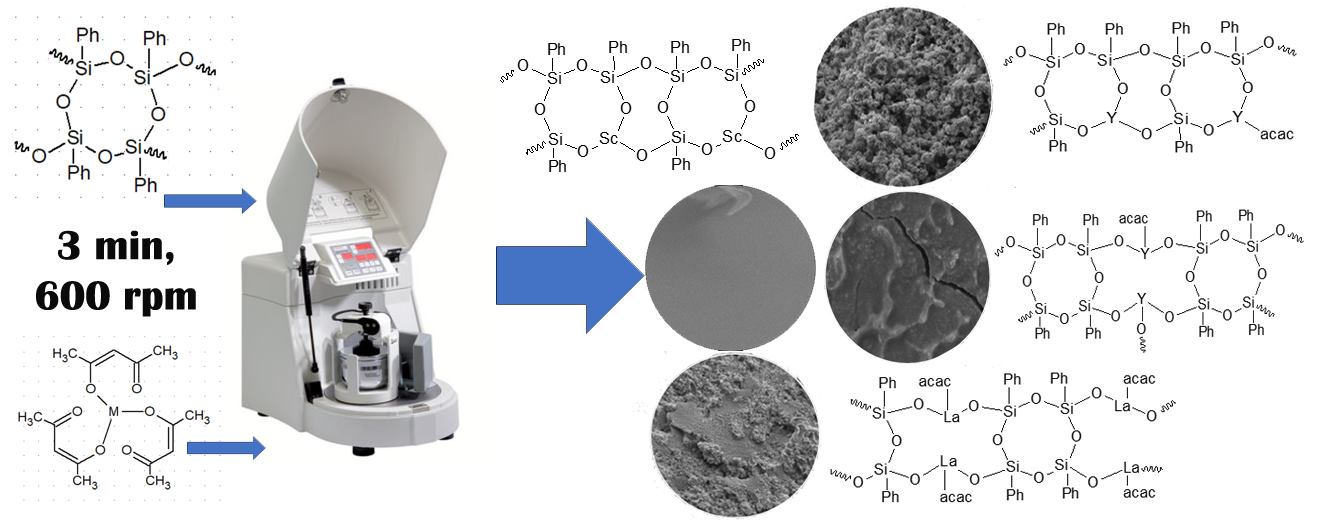
Graphical Abstract
1. Introduction
Polyorganosiloxanes form cross-linked networks with a large specific surface area, exhibiting operational capability over a wide temperature range, including harsh climatic, radiation, and highly aggressive environments [1]. Due to these properties, they possess immense potential as building blocks for various advanced materials. They can be used to create protective coatings, selective membranes, and high-capacity sorbents, and find applications in coordination chemistry, materials science, and catalysis [2,3]. Polyorganosiloxanes are hydrophobic materials, yet they also exhibit oleophobic and silylophilic properties, which makes them valuable in the cosmetics industry [4]. When these polymers incorporate ions of noble and rare-earth metals, highly active catalytic systems are formed, serving as catalyst supports. Combined with their large surface area, this makes them highly sought-after in this field [5,6,7].
Currently, siloxane polymers containing rare-earth metals in their structure are of great interest. For example, the incorporation of lanthanum into tris(hydroxymethyl)aminomethane polysiloxane has yielded a novel surfactant. This compound exhibits a critical micelle concentration that is 2–15 times lower than that of common soap-like surfactants and also demonstrates a high ability to reduce surface tension [8]. Siloxanes containing terbium and europium ions can be used in optoelectronic devices. They exhibit enhanced stability and thermal stability, display catalytic properties, and, when iron oxides are introduced, acquire magnetic properties [9]. Interesting properties are also exhibited by the complex formed through cross-linking of polydibenzoylmethane siloxane and europium phenylsiloxane. These compounds display intense red luminescence in the visible region, with the intensity increasing as the number of ligands rises. Thanks to the “antenna effect”, these ligands absorb light and transfer energy to the europium ion, drastically enhancing the emission brightness. At temperatures up to 150 °C and under prolonged heating, the polymers exhibit thermally stimulated self-healing, with the healing rate increasing as temperature rises. The materials are sensitive to ammonia vapors, opening up possibilities for their use as sensors [10]. Some oligomeric europium siloxanes efficiently absorb ultraviolet radiation while maintaining transparency below 15% [11]. Using polyphenylsilsesquioxanes as a matrix for Eu3+ ions allows the fabrication of robust and thermally stable optical waveguides and microresonators that enhance spontaneous emission [12,13]. Composites of polyphenylsilsesquioxanes with niobium exhibit high resistance to atomic oxygen. Their films do not crack and maintain integrity even at high atomic oxygen fluences. Moreover, they demonstrate high thermal and radiation stability [14].
As a relatively new branch of chemistry, mechanochemistry is gaining increasing attention and has become a fast-growing research field. Its use as a synthetic approach is expanding, which is reflected in the steady rise of publications and the formation of new research teams. The key driver of growing interest in mechanochemistry is its versatility: it enables solvent-free chemical reactions, substantially reducing waste, environmental impact, and production costs. Moreover, mechanochemical syntheses yield significantly higher product outputs than comparable solution reactions. Mechanochemical activation also shortens reaction times, improves stoichiometric control, and increases product selectivity [15]. A range of compounds can be prepared by mechanochemical synthesis, including peptides and organic polymers [16,17], pharmaceutical agents [18,19,20,21,22], and organic metal complexes [23,24,25,26].
Although numerous well-established methods for synthesizing organosilicon compounds exist, e.g., [27], they rely on organic solvents and typically take from several hours to several days to complete. In addition, solution-phase syntheses can give rise to unwanted side processes, such as reagent–solvent interactions and product decomposition during isolation. Developing and investigating solid-state methods for synthesizing both low- and high-molecular-weight organosilicon compounds, notably mechanochemical synthesis, remains a highly relevant challenge. In addition, mechanochemistry involving high-molecular-weight organoelement compounds, especially organosilicon polymers, is still poorly understood. There is only scant information on the activation and mechanical degradation of such polymers [28].
A novel synthesis of metalophenylsiloxanes bearing scandium, yttrium, and lanthanum atoms in the polymer chain has been achieved under mechanochemical activation, as demonstrated in this work.
2. Materials and Methods
2.1. General Procedures
The work employed commercial solvents, which were purified and dehydrated using standard methods. The resulting physical constants agreed with the literature values. The salts of metals used in the syntheses (grade “chemically pure”) were used without additional purification. In some syntheses, the salts were pre-dried to remove associated water.
2.2. Synthesis Procedures for Parent Compounds
2.2.1. Synthesis of Polyphenylsilsesquioxane (PPSSO)
PPSSO was prepared according to the procedure described in [29]. A high-molecular-weight compound of the composition [(C6H5)SiO1.5·0.17H2O]n was obtained with a yield of 93.7%. Foundalytical data (found/calculated, %): Si: 21.2/21.2; C: 54.5/54.5; [H2O]: 2.3/2.3. The molecular weight was determined to be M ≥ 7000, and the dispersion coefficient was Kd = 1.89.
2.2.2. Synthesis of Metal Acetylacetonate Complexes
Tris-acetylacetonate of scandium was prepared according to the procedure described in [30]. A colourless crystalline substance with a melting point of 187–188 °C was isolated. For the compound of composition Sc(acac)3, the following analytical data were obtained (found/calculated, %): Sc: 13.1/13.1; C: 53.0/52.6. The IR spectrum of the compound is presented in Table S1 and Figure S1 in the Supplementary materials.
Tris-acetylacetonate of yttrium was prepared according to the procedure described in [31]. A white crystalline substance with a melting point of 131 °C was isolated. For the compound of composition Y(acac)3·3H2O, the following analytical data were obtained (found/calculated, %): Y: 20.2/20.2; C: 41.0/40.9; [H2O] 12.2/12.3. The IR spectrum of the compound is presented in Table S1 and Figure S2 in the Supplementary materials.
Tris-acetylacetonate of lanthanum was prepared according to the procedure described in [32]. A colourless crystalline substance was isolated. For the compound of composition La(acac)3·3H2O, the following analytical data were obtained (found/calculated, %): La: 28.3/28.3; C: 36.7/36.7; [H2O] 11.0/11.0. The IR spectrum of the compound is presented in Table S1 and Figure S3 in the Supplementary materials.
2.3. General Procedure for the Mechanochemical Synthesis of Metal Phenylsiloxanes
All syntheses were carried out in a planetary ball mill “Pulverisette 6” at 600 rpm for 3 min. The ratio of the grinding media mass to the mass of the useful load was 1.8–2.0. Steel balls with a diameter of 0.8 cm and a mass of 4.05 g were used as grinding media. The initial molar ratio of Si/M in all syntheses was 1:1. 0.05 mol of PPSSO and an equal amount of the scandium complex (synthesis 1), yttrium (synthesis 2), and lanthanum (synthesis 3) complexes were loaded into the reactor. The elemental analysis data for the compounds obtained in syntheses 1–3 are presented in Table 1.
Table 1. Results of elemental analysis of the products obtained in syntheses 1–3.
|
№ М |
wfr, % |
Found/Calculated, % |
Yield, % |
||||||
|---|---|---|---|---|---|---|---|---|---|
|
Si |
M |
C |
[H2O] |
Si/M |
C/Si |
Si |
M |
||
|
1 Sc |
1 35.95 |
17.65Sc2O3·(PhSiO1.5)0.7(SiO2)0.3·81.23H2O |
1.97 |
69.73 |
|||||
|
0.7/0.7 |
39.6/39.6 |
1.3/1.3 |
- |
1:35.3 |
4.3 |
||||
|
2 64.05 |
[(PhSiO1.5)3(ScO1.5)0.9(ScOL)0.1·0.43H2O]n |
88.92 |
29.81 |
||||||
|
17.7/17.8 |
9.5/9.5 |
47.0/46.9 |
1.6/1.7 |
3.0 |
6.5 |
||||
|
2 Y |
1 61.31 |
YL3·2H2O |
2.03 |
67.16 |
|||||
|
0.2/- |
21.0/21.0 |
42.7/42.5 |
- |
- |
- |
||||
|
2 24.78 |
[(PhSiO1.5)2.5(YO1.5)0.52(YOL)0.48·0.94H2O]n |
57.92 |
23.01 |
||||||
|
14.1/14.1 |
17.8/17.9 |
42.1/42.1 |
3.4/3.4 |
2.5 |
6.96 |
||||
|
3 13.91 |
[(PhSiO1.5)4(YO1.5)0.44(YOL)0.56·1.4H2O]n |
36.44 |
9.14 |
||||||
|
15.8/15.9 |
12.6/12.6 |
45.3/45.6 |
3.6/3.7 |
4.0 |
6.7 |
||||
|
3 La |
1 47.32 |
LaL3·3.68H2O |
- |
47.21 |
|||||
|
- |
27.6/27.6 |
35.4/35.8 |
- |
- |
- |
||||
|
2 52.68 |
[(PhSiO1.5)1.92(LaOL)·2.55H2O]n |
92.57 |
48.36 |
||||||
|
9.8/9.8 |
25.3/25.4 |
36.0/36.2 |
8.4/8.4 |
1.92 |
10.28 |
||||
2.4. Methods for Separating Reaction Mixtures into Fractions
2.4.1. Work-Up of the Reaction Mixtures from Syntheses 1 and 2
After activation, the reaction mixtures were treated with warm water (35 °C) to dissolve the unreacted complex into solution. Once the mixture had been washed free of the complex, the residue was dissolved in chloroform and precipitated with hexane. The resulting compounds were dried in a vacuum oven at 75 °C until constant weight was achieved.
2.4.2. Work-Up of the Reaction Mixtures from Synthesis 3
After activation, the reaction mixtures were treated with ethanol to dissolve the unreacted complex into solution. Once the mixture had been washed free of the lanthanum complex, the residue was dissolved in chloroform and precipitated with hexane. The resulting compounds were dried in a vacuum oven at 75 °C until constant weight was achieved.
2.5. Methods of Analysis
2.5.1. Elemental and Functional Analysis
Decomposition of the compounds was carried out using the Kjeldahl and Schöniger methods (for carbon determination). Carbon was determined by the Terentiev–Luskina method, as well as using a Flash EA 1112CHN/MAS200 carbon, hydrogen, and nitrogen analyser (ThermoFinnigan MAT GmbH, San Jose, CA, USA). Silicon was determined by gravimetric analysis. Rare-earth metals were determined using an ICPE-9000 inductively coupled plasma atomic emission spectrometer (Shimadzu Corporation, Kyoto, Japan). Hydroxyl groups and water were determined by Karl Fischer titration.
2.5.2. Physical and Physicochemical Methods of Analysis
Gel Permeation Chromatography
Gel permeation chromatography (GPC) was performed on a 980 mm long column with a diameter of 12 mm, filled with a copolymer of polystyrene and 4% divinylbenzene. The diameter of the grains is 0.08–1 mm. The eluent was toluene, and the flow rate was 1 mL/min. The size of the sample was ~0.2 g. The detection was carried out by a gravimetric method according to the content of dry residue in the fractions. A portion of the substance was dissolved in 2 mL of toluene and passed through a column. Fractions of the solution were collected in 3 mL, and the solvent was removed in a drying cabinet. The column was preliminarily calibrated with substances with different molecular masses: polydimethylsiloxane H[Me2SiO]30OH (M = 2238), octaphenylcyclotetrasiloxane [Ph2SiO]4 (M = 792), hexaphenylcyclotrisiloxane [Ph2SiO]3 (M = 594), and benzoic acid (M = 122).
IR Spectroscopy
IR spectra were recorded on a Spectrum BX 400 FTIR spectrometer (Perkin Elmer, Waltham, MA, USA) in potassium bromide.
X-ray Phase Analysis
X-ray phase analysis was performed on a Bruker AXS D8 Advance X-ray diffractometer (Bruker, Ettlingen, Germany). Bregg-Brentano survey geometry, monochromator, Fe Kα-radiation, wavelength = 0.19373.
Scanning Electron Microscopy
The surface morphology, structure, and composition of the obtained compounds were studied using a Carl Zeiss Ultra 55+ scanning electron microscope (Carl Zeiss, Oberkochen, Germany). The spatial resolution was 1.0–4.0 nm at 15–0.1 kV. The magnification range was 12–900,000× in secondary electron mode and 100–900,000× in backscattered electron mode. The electron source was a field-emission type. The accelerating voltage range was 100–30,000 V. The operating current range was 4 pA–20 nA. Elemental surface analysis was performed using an Oxford Instruments X-MAX 80 energy-dispersive detector (Oxford Instruments, Abingdon, UK).
Thermogravimetric Analysis
The thermal effects and thermal characteristics of the studied substances and materials were determined using a DTG-60AH thermogravimetric/differential thermal analyser (Shimadzu, Kyoto, Japan). Air was used as the atmosphere in the applied methods.
Gas Chromatography
The determination of volatile substances and degradation products of the initial metal complexes was carried out using an Agilent 6890/5975B gas chromatograph with a quadrupole mass-selective detector (Agilent, Santa Clara, CA, USA).
3. Results and Discussion
Previously [33], we demonstrated that activation of PPSSO for 3 min leads to the formation of products with a range of molecular weights from 250 Da to 1500 Da. In contrast, activation of the initial boron difluoride acetylacetonate was primarily determined by air humidity and resulted in the degradation of 27% of the complex. The formation of low-molecular-weight degradation products of the starting compounds was confirmed by GC-MS, GPC, and MALDI-TOF methods. It was assumed that the interaction of rare-earth element acetylacetonates with PPSSO would also proceed via partial degradation of the complexes, followed by condensation of the resulting intermediate active species.
After activation of scandium tris-acetylacetonate with PPSSO (synthesis 1) and opening the reactor, a persistent odor of acetic acid was detected. After the reaction mixture had been treated with ethanol and the appropriate sample preparation steps completed, the solution was analysed via gas chromatography. Acetic acid, acetone, and acetylacetone were detected in the mixture (See Table S2, Figure S4, Figure S5, Figure S6, Figure S7, Figure S8, Figure S9, Figure S10, Figure S11, Figure S12, Figure S13, Figure S14 and Figure S15 in the supplementary materials). Upon treatment of the reaction mixture with warm water, the initial scandium acetylacetonate was not found. Treatment of the reaction mixture with chloroform resulted in the separation of two fractions: one insoluble in chloroform (fraction 1) and one soluble (fraction 2). Precipitation of fraction 2 with hexane did not lead to the formation of additional compounds. According to elemental analysis data (Table 1), fraction 1, with a mass fraction of 35.95%, is hydrated scandium oxide with a minor content of degradation products from the initial organosilicon polymer. Fraction 2 of the synthesis, according to elemental analysis and GPC data, is a high-molecular-weight compound (M ≥ 7000, see Figure S16 in the supplementary materials) with a Si/Sc ratio differing from the target value and equal to 3:1. The scandium conversion rate was 29.81%. GPC has revealed that the synthesis product contains no low-molecular-weight compounds, including the initial scandium complex (see Figure S17 in the supplementary materials). However, there is a low-intensity peak corresponding to a molecular weight of 4000, which indicates the formation of smaller macromolecules. Since the content of this fraction in the polymer is negligible, it was not isolated or analysed separately.
In the IR spectrum of fraction 2 (Figure 1), absorption bands characteristic of phenylsiloxanes are present (3013, 3051, 3074, 1595, 1431, 1134, 1028 cm−1). The spectrum also clearly shows absorption bands characteristic of vibrations of bonds in free silanol groups (3616 and 902 cm−1) and associated water (3408 and 1618 cm−1). Vibrations of Sc–O–Si bonds are overlapped by an intense absorption band of the siloxane bond. Of note is the splitting of the siloxane bond into two components at 1028 and 1066 cm−1, which may indicate an alternation of Si–O–Si and Si–O–Sc bonds, as well as an increase in the ring size.
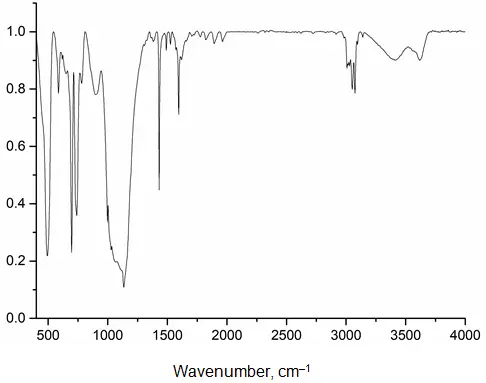
In contrast to the initial PPSSO, the X-ray diffraction pattern of the high-molecular-weight fraction exhibits a reflection indicative of a crystalline phase. Compared to the initial PPSSO (d1 = 11.605 Å, 2θ = 7.61°; d2 = 4.609 Å, 2θ = 19.24°), the interplanar spacing increases significantly (d1 = 12.620 Å, 2θ = 6.99°), which is attributed not only to the incorporation of a scandium atom into the polymer chain but also to the formation of larger cycles. At the same time, the intrachain distances remain virtually unchanged (d2 = 4.612 Å, 2θ = 19.23°). The appearance in the diffraction pattern of a high-molecular-weight fraction of a reflection characteristic of the crystalline phase (d3 = 6.707 Å, 2θ = 13.19°) is associated with the formation of a more ordered structure. This reflection is not characteristic of the initial scandium complex or its oxide, which rules out the formation of a mixture.
In the micrographs of scandium phenylsiloxanes (Figure 2a,b), it is evident that the polymer surface is homogeneous, with no globule’s characteristic of the initial PPSSO, as well as no cracks or chips. According to energy-dispersive analysis carried out at eight points, scandium is virtually absent on the surface (its content ranges from 0% to 0.03%), which appears to be due to the heteroatom being located inside the polymer chains. At the same time, the silicon content on the surface varies from 8.68% to 10.63%. The X-ray diffraction analysis data are in good agreement with the elemental analysis of the polymer surface.

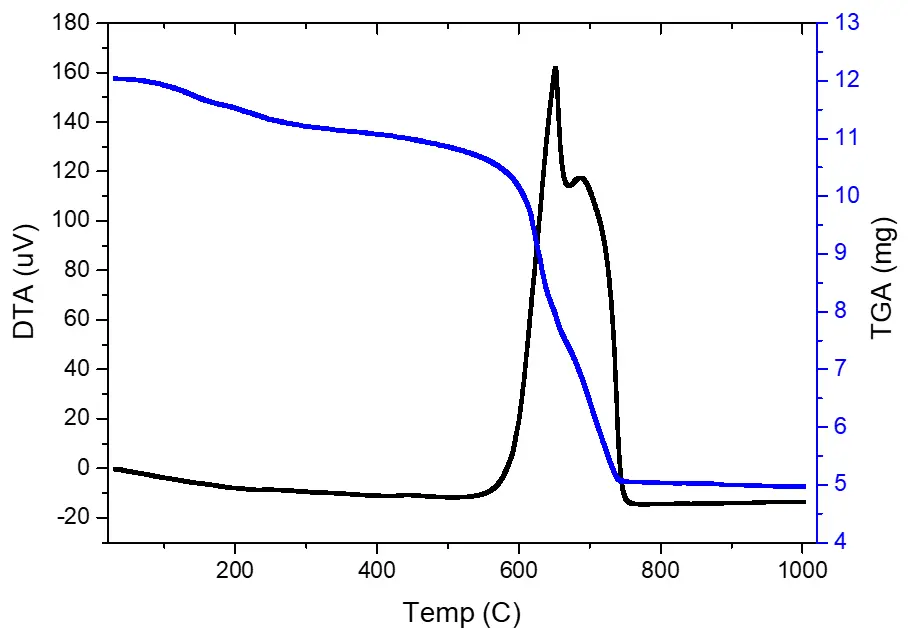
The high-molecular-weight product obtained in synthesis 1 is nearly identical to the initial PPSSO in terms of thermal stability (Figure 3). However, unlike phenylsiloxane, the DTA curve shows two exothermic peaks corresponding to the combustion of the organic portion of the molecules. The first exothermic effect occurs at a peak temperature of 650.74 °C, and the second, less intense one, at 687.38 °C. For the initial PPSSO, the organic portion burns off at 676.91 °C.
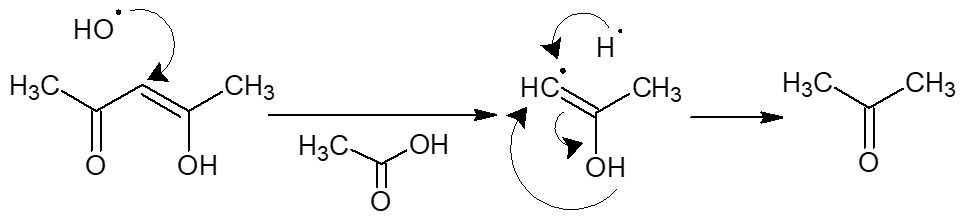
Based on the synthesis performed, it can be concluded that the scandium atom incorporated into the polymer chain exhibited catalytic activity toward the acetylacetone released during the activation process. The hydroxyl radical generated during activation (as demonstrated in our works [33,34,35]) attacks the molecule of the released acetylacetone. The rate of this process is very high due to diffusion as well as the catalytic properties of the scandium atom. This leads to the formation of acetone and acetic acid during the activation process:
According to the data from elemental analysis, IR spectroscopy, and X-ray diffraction analysis, the formula of the elementary unit of the resulting scandium phenylsiloxane is as follows:
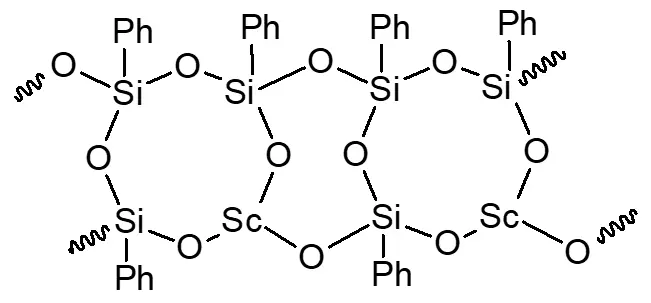
In synthesis 2, yttrium acetylacetonate was used as the initial complex. After activation and separation of the mixture, three fractions were isolated. The first fraction, soluble in warm water, was the original yttrium complex, with a yield of 61.31% (Table 1). The second and third fractions were high-molecular-weight products with Si/Y ratios differing from the target value: 2.5:1 for fraction 2 and 4:1 for fraction 3. Both fractions retained a considerable number of acetylacetonate groups. GPC has revealed that the synthesis product contains no low-molecular-weight compounds, including the initial yttrium complex (see Figure S18 in the supplementary materials). The IR spectra of fractions 2 and 3 are virtually identical (Figure 4, shown for fraction 2).
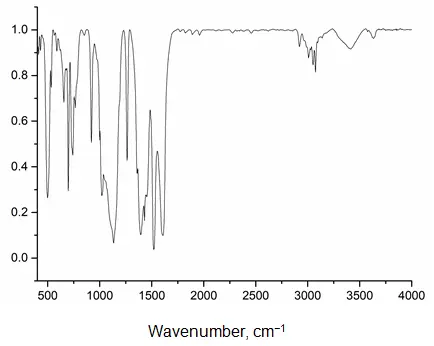
The spectra show absorption bands characteristic of bond vibrations in phenylsiloxanes (3007, 3051, 3074, 1431, 1134, 1020 cm−1), as well as in the acetylacetonate fragment (1608, 1520, 1394, 1359, 1263 cm−1). There are also low-intensity absorption bands characteristic of vibrations of silanol groups and associated water (3630, 850, and 3412 cm−1).
According to the X-ray diffraction analysis data, fraction 2 is an amorphous compound. It is worth noting that the maximum of the first reflection—which characterizes interchain distances—coincides in value with that of scandium phenylsiloxanes and exceeds the corresponding value for the initial PPSSO by more than 1 Å (d1 = 12.620 Å, 2θ = 6.99o). The second reflection (unlike that of scandiumphenylsiloxane) has a lower value than in the original organosilicon compound (d2 = 4.594 Å, 2θ = 19.30o). The compression of intrachain distances appears to be associated with the formation of coordination bonds between siloxane oxygen atoms and yttrium atoms.
Polyyttriumphenylsiloxane (fraction 2) differs from scandiumphenylsiloxane in terms of surface morphology (Figure 5a–d). The polymer has a rather developed surface featuring spheres of various sizes. Both intact and fractured globules are present on the surface (Figure 5d). The yttrium content on the surface ranges from 0.57% to 8.86%, while the silicon content varies from 3.07% to 7.46%.

Figure 5. Micrographs of fraction 2 from synthesis 2 at 200 nm (a), 1 μm (b), 2 μm (c), and 10 μm (d).
The surface of fraction 3 from synthesis 2 (Figure 6) differs from that of fraction 2. The surface of fraction 3 exhibits a layered structure (Figure 6b). When the magnification is increased to 200 nm, surface relief becomes visible (Figure 6a). Notably, there are no spheres characteristic of the original PPSSO that were partially retained in fraction 2.

The elemental composition of the polymer surface also differs significantly. Specifically: yttrium content ranges from 0.11% to 0.45%; silicon content ranges from 4.04% to 6.63%. The main elements constituting the surface of the fraction are: carbon (≥64.4%); oxygen (≥21.6%).
The DTA curves also reveal differences between fractions 2 and 3. Specifically, the DTA curve of fraction 3 (Figure 7, 1) shows three distinct maxima corresponding to exothermic effects. Each effect is accompanied by mass loss, which we attribute to the following processes:
-
At the peak corresponding to 199.4 °C, polymer dehydration occurs—i.e., the release of water molecules associated with the metal.
-
The effects at 363.8 °C and 630.2 °C correspond to the combustion of: acetylacetonate groups bonded to the metal atom; phenyl substituents attached to silicon atoms. The thermal stability of the resulting polymer is slightly lower than that of the initial PPSSO (676.9 °C).
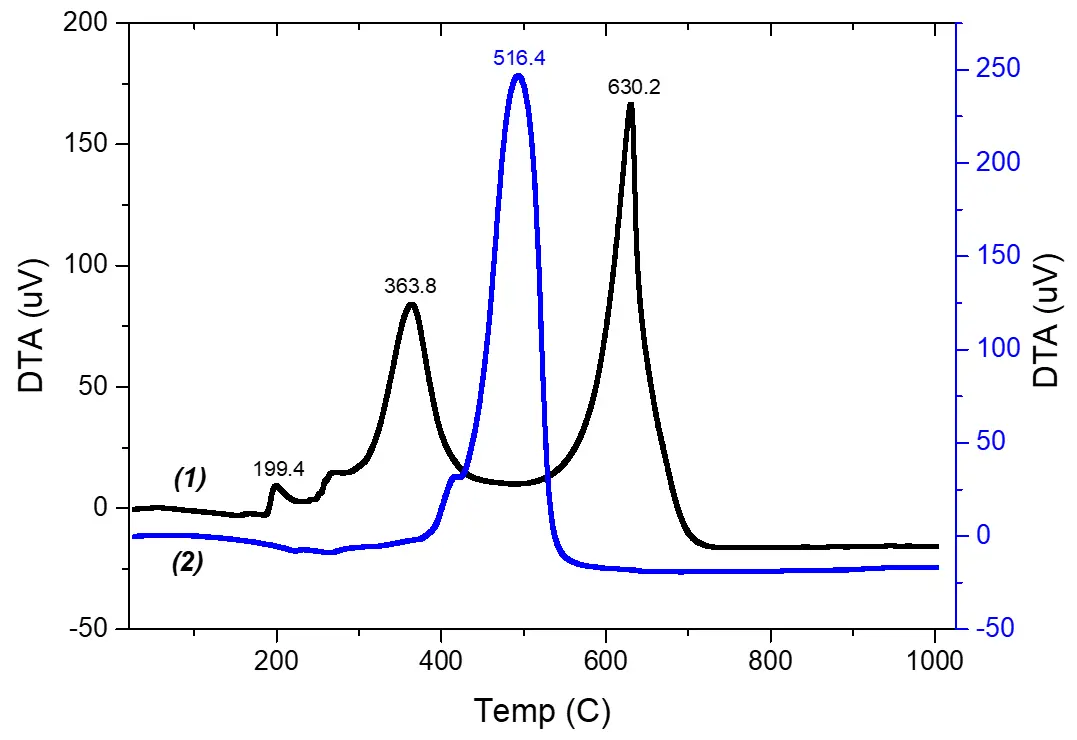
The data obtained by DTA-TGA are consistent with the results of elemental analysis as well as with surface analysis. In contrast to fraction 3, the DTA curve of fraction 2 (Figure 7, 2) shows only one clearly defined exothermic effect, which occurs at a peak temperature of 516.4 °C and corresponds to the combustion of the organic part of the polymer. The thermal stability of the resulting compound is noticeably lower than that of fraction 3 and significantly lower than that of the initial PPSSO. This difference appears to be related to: the catalytic properties of the yttrium atom; the maximally developed surface area of the resulting polymer (Figure 5a–d).
According to the data from chemical and instrumental analysis methods, Fraction 2 is a polymer product containing structural units 1, and Fraction 3 is a polymer product containing structural units 2:
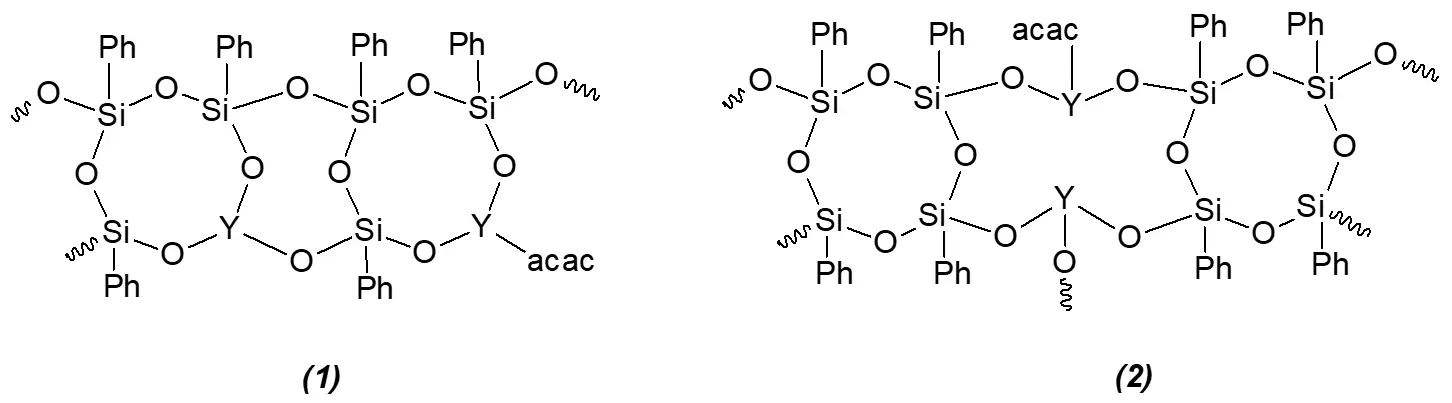
The separation of the reaction mixture obtained via mechanochemical activation of PFSCO with lanthanum acetylacetonate (synthesis 3) yielded two fractions. According to elemental analysis and IR spectroscopy data, fraction 1 from synthesis 3 is the unreacted hydrated lanthanum complex.
Fraction 2, according to elemental analysis (Table 1) and GPC, is a high-molecular-weight lanthaniumphenylsiloxane with a relative molecular mass of M ≥ 7000. GPC has revealed that the synthesis product contains no low-molecular-weight compounds, including the initial lanthanum complex (see Figure S19 in the supplementary materials). The yield of the high-molecular-weight fraction was 52.68%, and the degree of lanthanum incorporation into the polymer chain was 48.36%. The resulting Si/La ratio differs almost twofold from the intended ratio (1.92:1). Precipitation of the toluene solution of the resulting compound with heptane did not lead to the formation of additional fractions.
In the IR spectrum of fraction 2 (Figure 8), absorption bands characteristic of phenylsiloxanes are present at 3007, 3051, 3074, 1595, 1431, 1134, and 1028 cm−1, as well as bands corresponding to acetylacetonate fragments at 1520, 1385, and 1261 cm−1.
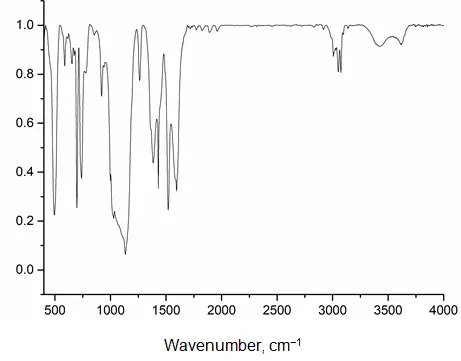
In the IR spectrum of fraction 2, apart from the absorption bands characteristic of phenylsiloxanes and acetylacetonate groups, minor absorption bands are also present. These correspond to: vibrations of free silanol group bonds (at 3620 and 854 cm−1) and associated water (at 3422 cm−1). The elemental analysis and IR spectroscopy data are in good agreement with each other.
Compared to the initial PPSSO, the first reflection maximum in the diffraction pattern shifts to 2θ = 6.92°, with an increase in the interplanar spacing from 11.605 to 12.756 Å. In contrast, the intrachain distance slightly decreases (from 4.609 to 4.491 Å). X-ray diffraction data indicate that the lanthanum atom has entered the interchain space. The decrease in intrachain distances is attributed to the “pulling” of siloxane oxygen atoms toward the lanthanum atom.
The surface of the obtained polymer is rather developed, yet it lacks pores and globules (Figure 9). According to EDX data, the lanthanum content on the polymer surface ranges from 0.11% to 0.45%, while the silicon content varies from 4.04% to 6.63%. The main component of the surface is the organic framework, with carbon content ranging from 64.4% to 66.2%.

Thermogravimetric analysis also confirms that all the carbon is concentrated on the polymer surface. The thermal stability of the polymer is only slightly different from that of the initial PPSSO, and the sole exothermic process maximum is observed at 665.12 °C.
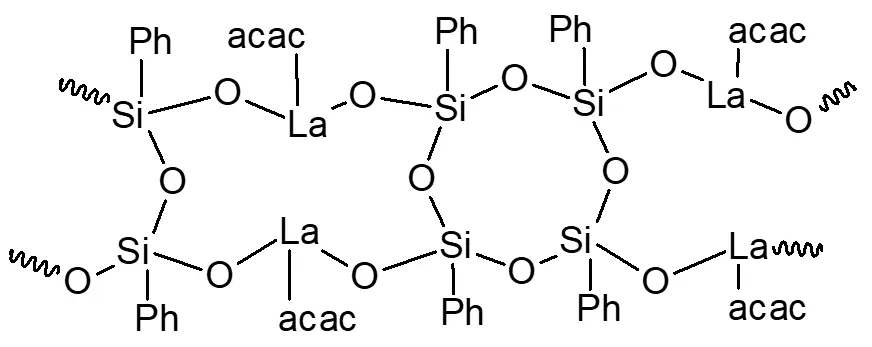
Based on the synthesis performed, we can hypothesize that the structural unit of the polymer obtained in synthesis 3 is represented by the following formula:
4. Conclusions
Thus, we have for the first time carried out mechanochemical syntheses based on polyphenylsilsesquioxane and acetylacetonates of certain rare-earth metals.
The fundamental possibility of synthesizing element-phenylsiloxanes containing scandium, yttrium, and lanthanum atoms in the chain has been demonstrated. Based on the syntheses performed, a certain pattern can be observed in the incorporation of rare-earth elements into the siloxane polymer chain:
-
-
As the ionic radius increases, the ability of the acetylacetonate complex surface to donate an electron pair to the adsorbed acid (i.e., the silsesquioxane surface) also increases.
-
-
The stability of acetylacetonate complexes decreases in the series Sc–Y–La, which also facilitates more complete incorporation of the heteroatom into the polymer chain under mechanical action.
-
-
The degree of heteroatom conversion increases in the series Sc–Y–La.
None of the syntheses based on rare-earth metal acetylacetonates yielded polymers with the target silicon-to-metal ratio. However, a tendency was observed for the organic ligand to be retained at the metal atom.
A specific feature of the synthesis based on scandium acetylacetonate is that the insoluble fraction did not contain the initial metal complex. Instead, a complete detachment of the organic group from the scandium atom occurred, resulting in the formation of an insoluble oxide.
The presence of partially preserved silanol groups in the resulting scandium-, yttrium-, and lanthanum-containing phenylsiloxanes, as well as water molecules within the polymers, suggests that the metal incorporation process predominantly proceeded via a radical mechanism involving the cleavage of siloxane bonds.
Supplementary Materials
The following supporting information can be found at: https://www.sciepublish.com/article/pii/1073, Table S1: Assignment of absorption bands in the IR spectra of acetylacetonate complexes of scandium, yttrium and lanthanum; Figure S1: IR spectrum of scandium tris-acetylacetonate; Figure S2: IR spectrum of yttrium tris-acetylacetonate; Figure S3: IR spectrum of lanthanum tris-acetylacetonate; Table S2: Composition of the ethanol-extracted reaction mixture; Figure S4: Gas chromatogram of the ethanol-extracted reaction mixture; Figure S5: Mass spectrum of Carbon dioxide; Figure S6: Mass spectrum of Acetone; Figure S7: Mass spectrum of Ethanol; Figure S8: Mass spectrum of 2,3-Butanedione; Figure S9: Mass spectrum of Ethyl Acetate; Figure S10: Mass spectrum of Acetic acid; Figure S11: Mass spectrum of Toluene; Figure S12: Mass spectrum of 2,4-Pentanedione; Figure S13: Mass spectrum of 2-oxo-ethyl ester of Propanoic acid; Figure S14: Mass spectrum of 1-(acetyloxy)-2-Propanone; Figure S15: Mass spectrum of Ethyl 2-acetoxy-2methylacetoacetate; Figure S16: Chromatograms of the initial PPSSO and the products of syntheses 1–3; Figure S17: Chromatogram of the products from synthesis 1 based on scandium acetylacetonate; Figure S18: Chromatogram of the products from synthesis 1 based on yttrium acetylacetonate; Figure S19: Chromatogram of the products from synthesis 1 based on lanthanum acetylacetonate.
Acknowledgments
The authors express their deep gratitude to the Molecular Analysis Laboratory at the Institute of High-Tech Technologies and Advanced Materials (Far Eastern Federal University), as well as to Natalya Valeryevna Maslova, Engineer at the Department of Chemistry and Materials, for conducting several chemical analysis methods.
Author Contributions
Conceptualization, V.L. and A.K.; Methodology, V.L., I.K. and V.T.; Software, V.L.; Validation, V.L., A.K., I.K. and V.T.; Formal Analysis, V.L., I.K. and V.T.; Investigation, V.L., A.K., I.K. and V.T.; Resources, V.L. and A.K.; Data Curation, V.L. and A.K.; Writing—Original Draft Preparation, V.L.; Writing—Review & Editing, V.L. and A.K.; Supervision, V.L. and A.K.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data supporting the results of this study are contained in the supplementary materials to the article, available at: www.sciepublish.com/xxx/s1. Additional data, including raw files obtained during the study using instrument-specific software, as well as author-related information, may be accessed upon request to the corresponding author. Requests will be reviewed on a case-by-case basis, subject to applicable restrictions.
Funding
This research received no external funding.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Raygorodsky I, Kopylov V, Kovyazin A. Membrane Materials for Gas and Vapor Separation; Wiley: Hoboken, NJ, USA, 2017; pp. 17–51. [Google Scholar]
- Pielichowski K, Njuguna J, Janowski B, Pielichowski J. Supramolecular Polymers Polymeric Betains Oligomers. Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 2006; Volume 201, pp. 225–296. DOI:10.1007/12_077 [Google Scholar]
- Khoroshavina JV, Frantsuzova JV, Nikolaev GA. The properties of vulcanisates based on a polyphenylsilsesquioxane-polydimethylsiloxane block copolymer. Int. Polym. Sci. Technol. 2015, 42, 13–16. DOI:10.1177/0307174x1504200903 [Google Scholar]
- O’Lenick AJ. Silicone Polymers: New Possibilities in Nanotechnology. ACS Symp. Ser. 2007, 961, 165–175. DOI:10.1021/bk-2007-0961.ch009 [Google Scholar]
- Anisimov AA, Minyaylo EO, Shakirova AR, Shchegolikhina OI. Evolution of Organometallasiloxanes. Polym. Sci. 2023, 65, 230–258. DOI:10.1134/S181123822370042X [Google Scholar]
- Lindner E, Schreiber R, Schneller T, Wegner P, Mayer HA, Göpel W, et al. Synthesis of Polysiloxane-Bound (Ether-phosphine)palladium Complexes. Stoichiometric and Catalytic Reactions in Interphases. Inorg. Chem. 1996, 35, 514–525. DOI:10.1021/ic950412m [Google Scholar]
- Lu Z, Lindner E, Mayer HA. Applications of sol-gel-processed interphase catalysts. Chem. Rev. 2002, 102, 3543−3578. DOI:10.1021/cr010358t [Google Scholar]
- Racles C, Silion M, Iacob M. Lanthanum complex of a multifunctional water-soluble siloxane compound—Synthesis, surface activity and applications for nanoparticles stabilization. Colloids Surf. A Physicochem. Eng. Asp. 2014, 462, 9–17. DOI:10.1016/j.colsurfa.2014.08.016 [Google Scholar]
- Feng J, Zhang H. Hybrid materials based on lanthanide organic complexes: A review. Chem. Soc. Rev. 2013, 42, 387–410. DOI:10.1039/c2cs35069f [Google Scholar]
- Kim EE, Kononevich YN, Dyuzhikova YS, Ionov DS, Khanin DA, Nikiforova GG, et al. Cross-Linked Luminescent Polymers Based on β-Diketone-Modified Polysiloxanes and Organoeuropiumsiloxanes. Polymers 2022, 14, 2554. DOI:10.3390/polym14132554 [Google Scholar]
- Kim EE, Ershova TO, Belova AS, Khanin DA, Bashkova EV, Nikiforova GG, et al. Luminescent Composite Films Based on Mechanically Strong Ladder-like Polyphenylsilsesquioxane and Oligophenyleuropiumsiloxane. Chin. J. Polym. Sci. 2024, 42, 1793–1801. DOI:10.1007/s10118-024-3190-9 [Google Scholar]
- Hasegawa Y, Kawai H, Nakamura K, Yasuda N, Wada Y, Yanagida S. Molecular design of luminescent Eu(III) complexes as lanthanide lasing material and their optical properties. J. Alloys Compd. 2006, 408–412, 669–674. DOI:10.1016/j.jallcom.2004.12.145 [Google Scholar]
- Manseki K, Hasegawa Y, Wada Y, Yanagida S. Photophysical properties of tetranuclear Eu(III) complexes in polyphenylsilsesqioxane (PPSQ). J. Alloys Compd. 2006, 408–412, 805–808. DOI:10.1016/j.jallcom.2005.01.071 [Google Scholar]
- Andropova US, Aysin RR, Serenko OA, Ershova TO, Anisimov AA, Chernik VN. Ladder Polyphenylsilsesquioxanes and Their Niobium-Siloxane Composite as Coating Materials: Spectroscopy and Atomic Oxygen Resistance Study. Polymers 2023, 15, 3299. DOI:10.3390/polym15153299 [Google Scholar]
- Howard JL, Cao Q, Browne DL. Mechanochemistry as an emerging tool for molecular synthesis: What can it offer? Chem. Sci. 2018, 9, 3080–3094. DOI:10.1039/c7sc05371a [Google Scholar]
- Friscic T, Mottillo C, Titi HM. Mechanochemistry for Synthesis. Angew. Chem. Int. Ed. 2020, 59, 1018–1029. DOI:10.1002/anie.201906755 [Google Scholar]
- Hu L, Xu S, Zhao Z, Yang Y, Peng Z, Yang M, et al. Ynamides as Racemization-Free Coupling Reagents for Amide and Peptide Synthesis. J. Am. Chem. Soc. 2016, 138, 13135–13138. DOI:10.1021/jacs.6b07230 [Google Scholar]
- Atapalkar RS, Kulkarni AA. Batch and continuous flow mechanochemical synthesis of organic compounds including APIs. React. Chem. Eng. 2024, 9, 10–25. DOI:10.1039/D2RE00521B [Google Scholar]
- Arkhipov IA, Dushkin AV, Khalikov SS, Varlamova AI, Arisov MV, Glamazdin II, et al. The effect of mechanochemical technology on the anthelmintic efficacy of solid dispersion of albendazole. Biopharm. J. 2021, 13, 36–41. Available online: https://www.researchgate.net/publication/351214810_Vlianie_mehanohimiceskoj_tehnologii_na_antigelmintnuu_effektivnost_tverdoj_dispersii_albendazola (accessed on 11 February 2026). (In Russian)
- Khalikov SS, Dushkin AV. Strategies for solubility enhancement of anthelmintics (Review). Pharm. Chem. J. 2020, 54, 504–508. DOI:10.1007/s11094-020-02229-4 [Google Scholar]
- Wei W, Evseenko VI, Khvostov MV, Borisov SA, Tolstikova TG, Polyakov NE, et al. Solubility, Permeability, Anti-Inflammatory Action and In Vivo Pharmacokinetic Properties of Several Mechanochemically Obtained Pharmaceutical Solid Dispersions of Nimesulide. Molecules 2021, 26, 1513. DOI:10.3390/molecules26061513 [Google Scholar]
- Harris N, Benedict J, Dickie DA, Pagola S. Mechanochemical synthesis insights and solid-state characterization of quininium aspirinate, a glass-forming drug-drug salt. Acta Crystallogr. Sect. C Struct. Chem. 2021, 77, 566–576. DOI:10.1107/S2053229621008275 [Google Scholar]
- Boyde NC, Rightmire NR, Bierschenk EJ, Steelman GW, Hanusa TP, Brennessel WW. Reaction environment and ligand lability in group 4 Cp2MXY (X, Y = Cl, OtBu) complexes. Dalton Trans. 2016, 45, 18635–18642. DOI:10.1039/c6dt03199d [Google Scholar]
- Wenger LE, Hanusa TP. Synthesis without solvent: Consequences for mechanochemical reactivity. Chem. Commun. 2023, 59, 14210–14222. DOI:10.1039/D3CC04929A [Google Scholar]
- Leonardi M, Villacampa M, Menéndez JC. Multicomponent mechanochemical synthesis. Chem. Sci. 2018, 9, 2042–2064. DOI:10.1039/C7SC05370C [Google Scholar]
- Takahashi R, Gao P, Kubota K, Ito H. Mechanochemical protocol facilitates the generation of arylmanganese nucleophiles from unactivated manganese metal. Chem. Sci. 2023, 14, 499–505. DOI:10.1039/d2sc05468j [Google Scholar]
- Levitskii MM, Smirnov VV, Zavin BG, Bilyachenko AN, Rabkina AY. Metalasiloxanes: new structure formation methods and catalytic properties. Kinet. Catal. 2009, 50, 490–507. DOI:10.1134/S0023158409040041 [Google Scholar]
- Chen Y, Mellot G, van Luijk D, Creton C, Sijbesma RP. Mechanochemical tools for polymer materials. Chem. Soc. Rev. 2021, 50, 4100–4140. DOI:10.1039/d0cs00940g [Google Scholar]
- Kapustina AA, Libanov VV, Shapkin NP, Pobozhev KV. The study of the interaction of aluminum acetylacetonate with polyphenylsilsesquioxane with the help of mechanochemical activation. Izv. Vyssh. Uchebn. Zaved. Khim. Khim. Tekhnol. 2022, 65, 59–66. DOI:10.6060/ivkkt.20226512.6660 [Google Scholar]
- Anderson TJ, Neuman MA, Melson GA. Coordination Chemistry of Scandium. V. Crystal and Molecular Structure of tris(Acetylacetonato)scandium(III). Inorg. Chem. 1973, 12, 927–930. DOI:10.1021/ic50122a046 [Google Scholar]
- Cunningham JA, Sands DE, Wagner WF. Crystal and Molecular Structure of Yttrium Acetylacetonate Trihydrate. Inorg. Chem. 1967, 6, 499–503. DOI:10.1021/ic50049a014 [Google Scholar]
- Gavrilenko VV, Chekulaeva LA, Savitskaya IA, Garbuzova IA. Synthesis of yttrium, lanthanum, neodymium, praseodymium, and lutetium alkoxides and acetylacetonates. Russ. Chem. Bull. 1992, 41, 1957–1959. DOI:10.1007/BF00863354 [Google Scholar]
- Libanov V, Kapustina A, Shapkin N, Puzyrkov Z, Dmitrinok P. Mechanochemical synthesis of Polyboronphenylsiloxanes. Polymer 2020, 194, 122367. DOI:10.1016/j.polymer.2020.122367 [Google Scholar]
- Kapustina AA, Shapkin NP, Libanov VV. Preparation of polyboronphenylsiloxanes by mechanochemical activation. Russ. J. Gen. Chem. 2014, 84, 1320–1324. DOI:10.1134/S1070363214070123 [Google Scholar]
- Libanov VV, Kapustina AA, Shapkin NP, Rumina AA. Mechanochemical interaction of boron difluoride acetylacetonate with organosilicon derivatives of different functionality. Silicon 2019, 11, 1489–1495. DOI:10.1007/s12633-018-9969-y [Google Scholar]