Zoonotic Microsporidia: Host Regulation and Pathogenesis

Zoonotic Microsporidia: Host Regulation and Pathogenesis
Lingkang Liu-Tang
1
Jiaying Jiang
1
Miao Zhang
1
Qiaoling Huang
1
Maoshuang Ran
1
Jie Chen
1
Zeyang Zhou
1,2
Jialing Bao
1,*

Received: 24 April 2026 Revised: 18 May 2026 Accepted: 04 June 2026 Published: 08 June 2026

© 2026 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
1. Introduction
Microsporidia are a diverse group of obligate intracellular unicellular eukaryotic parasites. To date, approximately 220 genera and over 1700 species of microsporidia have been taxonomically identified [1,2]. These pathogens are widely distributed throughout natural ecosystems, and their host range encompasses nearly all animal groups, from invertebrates to vertebrates. Among the diverse microsporidian taxa, zoonotic microsporidia exhibit an exceptionally broad host tropism, representing a dual threat to public health security and agricultural economic sustainability [3]. These zoonotic pathogens can cause mild to severe gastrointestinal, neurological, and systemic clinical diseases, especially in those of compromised immunities such as AIDS patients, organ transplant recipients, and other immunosuppressed populations. Zoonotic microsporidia also infect domesticated animals such as cattle, swine, and poultry, resulting in substantial production losses and economic damage to the livestock industry [4,5,6]. Notably, species such as Encephalitozoon hellem, Encephalitozoon cuniculi, and Enterocytozoon bieneusi are the most prevalent and clinically relevant zoonotic microsporidian species worldwide.
Microsporidia spores can live outside of a host cell and in a natural environment for a long time because of the rigid, chitin-rich spore wall. When chances permit, the spores could start to invade the host. Infection begins when the polar filament extrudes, injecting the infectious sporoplasm directly into the cytoplasm of a host cell [7,8]. The subsequent developmental stages—merogony (vegetative proliferation) and sporogony (spore formation)—take place within specialized parasitophorous vacuoles in the host cytoplasm, allowing the parasite to complete its life cycle [9,10]. Like other well-known fungal pathogens such as Candida albicans and Cryptococcus neoformans, microsporidia do not remain passive during intracellular infection. Instead, these obligate intracellular parasites actively reprogram host cell functions to promote their own proliferation, colonization, and immune evasion [11,12]. Recent accumulating evidence shows that this host-modulatory activity spans multiple critical cellular layers, including host metabolic homeostasis, intracellular signaling transduction, innate and adaptive immune responses, and epigenetic modification [13]. Specifically, at the metabolic level, microsporidia hijack and reprogram host carbohydrate, lipid, and protein metabolic pathways to obtain essential nutrients and energy for their own biosynthesis [14]. At the signaling level, they suppress host immune defenses by interfering with key cascades such as the mitogen-activated protein kinase (MAPK) pathway, thereby enhancing their intracellular survival [15]. At the immune level, microsporidia evade host clearance through various strategies, including escaping autophagic degradation and inhibiting host cell apoptosis [16,17]. At the epigenetic level, mechanisms such as histone post-translational modifications and DNA methylation have been implicated in microsporidia-driven remodeling of host cell fate and function [18].
Despite considerable progress in understanding microsporidia–host interactions over the past decades, few systematic reviews have specifically focused on how zoonotic microsporidia modulate host cells. Moreover, the synergistic regulatory mechanisms that coordinate different dimensions of host manipulation remain poorly understood.
This review examines how zoonotic microsporidia regulate host cells following successful infection, focusing on four core areas: metabolic reprogramming, perturbations of key signaling pathways, immune evasion strategies, and epigenetic modifications. In addition, we compare these mechanisms with the host manipulation strategies employed by other well-characterized fungal pathogens, including Candida albicans and Cryptococcus neoformans. Our goal is to systematically summarize current knowledge, consolidate existing findings on the intracellular parasitic mechanisms of zoonotic microsporidia, and provide a solid theoretical foundation for future mechanistic studies and the development of novel preventive and therapeutic approaches against microsporidiosis.
2. Metabolic Modulation
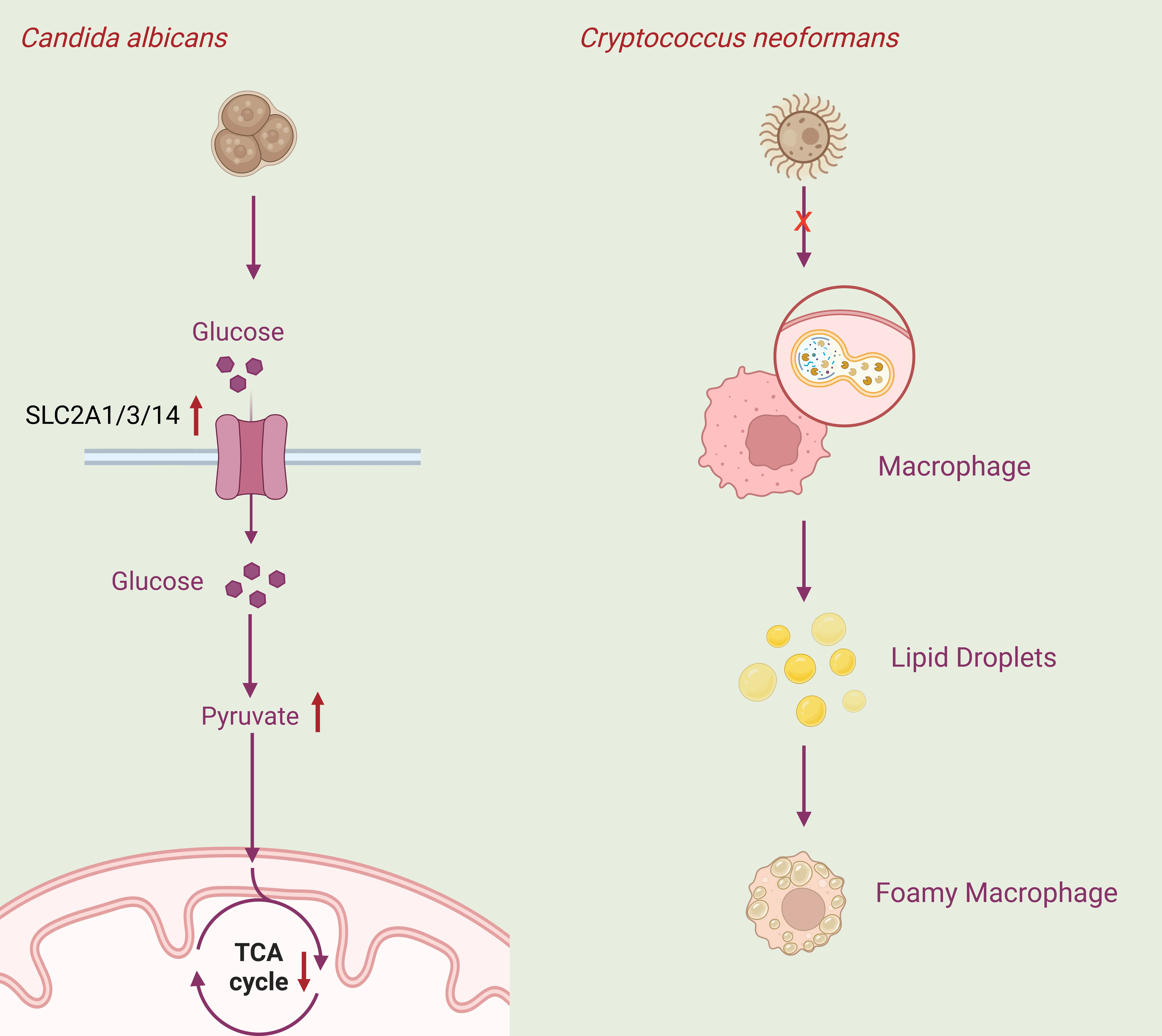
A key aspect of host manipulation begins with how these pathogens hijack fundamental metabolic pathways to secure energy and building blocks. Fungal pathogens, such as Cryptococcus neoformans, Candida albicans, and Aspergillus fumigatus, have the ability to regulate host metabolism to ensure their own proliferation (Figure 1A). Candida albicans infects host epithelial cells and regulates the cells’ uptake of glucose, thereby increasing the rate of glycolysis. At the same time, the activity of the host cells’ citric acid cycle decreases, creating an environment conducive to the fungus’s survival [19]. In the meantime, Cryptococcus neoformans inhibits the autophagy process in host cells during infection, leading to abnormal intracellular lipid accumulation and the formation of lipid-rich foam cells, which provide a microenvironment conducive to the pathogen’s nutrition and replication [20].
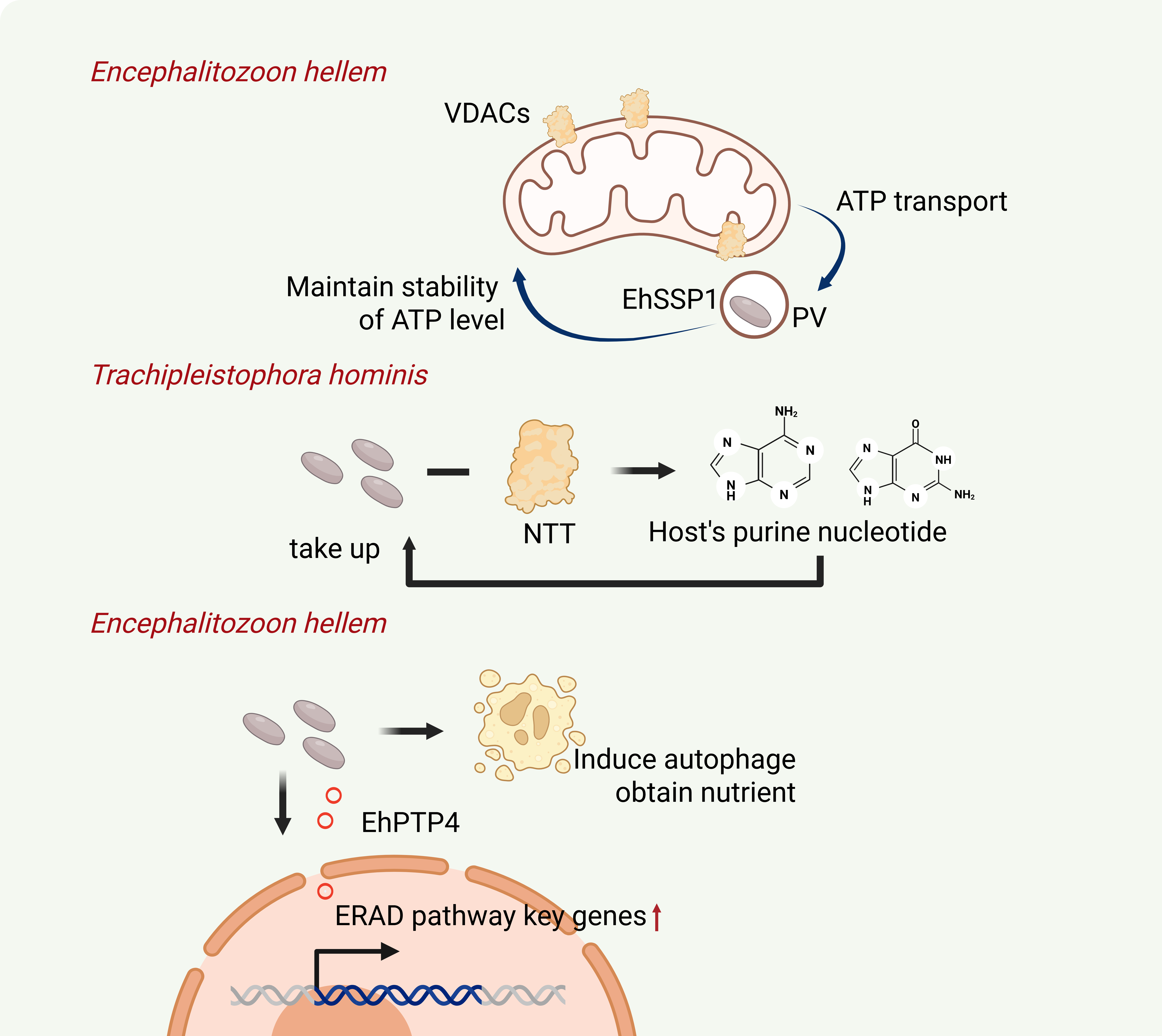
Most microsporidia only possess a tiny remnant mitochondrion (mitosome), which lost typical mitochondria cristae, organellar genome, and most canonical functions [21]. Therefore, the absence of certain biosynthetic pathways and genes involved in the citric acid cycle results in a strong dependence on the host. Once microsporidia have successfully invaded a host cell, their own energy and material reserves are far from sufficient for normal reproduction. They must compensate for their deficiencies in nucleotide synthesis and DNA replication by stealing nucleotides from the host, and they must also steal ATP from the host to meet their own energy needs. (Figure 1B). For instance, microsporidian Trachipleistophora hominis has been reported to take up host purine nucleotides via the nucleotide transporter NTT to compensate for its own deficiencies in nucleotide synthesis and DNA replication [22].
As for ATP intake,the E. hellem sporoplasm surface protein 1 (EhSSP1) has been found to interact with the host mitochondrial outer membrane protein, the voltage-dependent anion channels (VDACs). They can mediate the tight interaction between the host mitochondria and the parasitophorous vacuole (PV). This contact may facilitate the transportation of ATP from the host to the parasite, thereby meeting the energy requirements of the microsporidian [23]. Interestingly, microsporidia can maintain the host’s ATP levels at a steady state, ensuring the host cell’s survival and their own proliferation [24].
So how is this achieved? Based on existing research, we know that one way is to exploit the host’s autophagy. Unlike fungal pathogens such as Cryptococcus neoformans, which can suppress host autophagy, microsporidia, such as Encephalitozoon cuniculi, strongly induce autophagy in RK-13 cells, yet evade host-mediated degradation and utilize host autophagy as a nutrient source [25]. Although the mechanism by which microsporidia induce autophagy has not yet been fully elucidated, one relatively well-defined pathway involves enhancing host autophagy by increasing host ubiquitination. EHPTP4 is a polar tube protein of Encephalitozoon hellem. The N-terminus of the EhPTP4 protein contains a signal peptide, while the C-terminus contains a histidine-rich domain (HRD) and a nuclear localization signal (NLS). In both infected and transfected cells, EhPTP4 is secreted into the host cell nucleus. EhPTP4 significantly upregulates the expression of key genes (PDIA4, HERP, HSPA5, and Derlin3) involved in the host endoplasmic reticulum–associated degradation (ERAD) pathway and enhances the ubiquitination of host proteins, indicating that EhPTP4 functions in the regulation of host ERAD [25,26,27].
While metabolic modulation secures nutrient supply, the precise regulation of host signaling pathways may ensure intracellular pathogens a more non‑lytic, replication‑friendly environment. Therefore, the next section shifts focus to how microsporidia precisely manipulate key signaling pathways for immune evasion and survival.
 |
 |
|
(A) |
(B) |
Figure 1. Modulation on host metabolism by microsporidia and other fungal pathogens. (A) Candida albicans infection increases glycolytic activity in epithelial cells and reduces TCA cycle activity; Cryptococcus neoformans infection inhibits the function of host phagocytes, leading to abnormal intracellular accumulation of lipids. (B) The microsporidian E. hellem hijacks the host’s mitochondria and steals ATP to compensate for its own energy deficit; The microsporidian Trachipleistophora hominis hijacks the host nucleotide transporter NTT to take up host purine nucleotides to compensate for its own defects in nucleotide synthesis and DNA replication; the microsporidian E. hellem secretes the host nuclear localization protein EhPTP4 to activate key genes in the host ERAD pathway (VDACs: host mitochondrial outer membrane proteins, voltage-dependent anion channels; EhSSP1: spore surface protein 1 of E. hellem; NTT: nucleotide transporter; EhPTP4: secretory protein of E. hellem, localized to the host cell nucleus).
3. Signaling Pathway Fine-Tuning
It is very impressive that pathogens such as microsporidia, despite having highly reduced genomes, still possess numerous genes encoding host-manipulating molecules, such as serpins, ricin-like proteins, and protein phosphatases [15,28,29,30]. To this perspective, microsporidia are similar to typical fungal pathogens such as Cryptococcus neoformans and Candida albicans, which can manipulate host signaling pathways through effector proteins and cell wall components to achieve immune evasion, intracellular colonization, and proliferation.
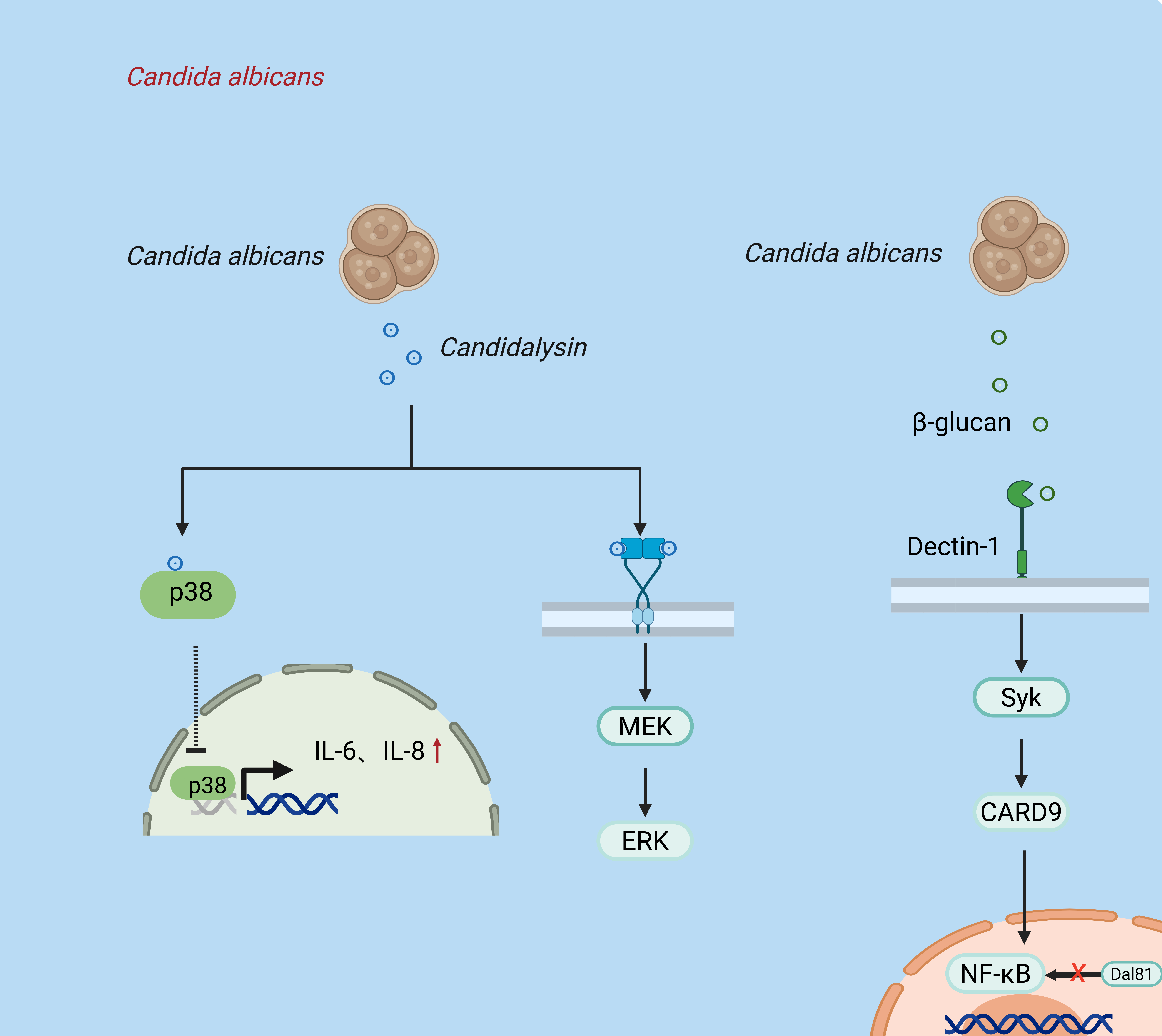
Different effectors targeting at different points of the signaling pathway may result in various cellular responses (Figure 2). For instance, candidalysin, the key toxin of Candida albicans, can activate the p38 MAPK pathway in epithelial cells, inducing the release of pro-inflammatory factors such as IL-6 and IL-8 [31]. Interestingly, candidalysin can also transactivate the EGFR-ERK pathway, precisely regulating the balance between cell damage and repair, thereby providing crucial support for hyphal invasion and tissue colonization [32]. In addition, the β-glucan in the cell wall of Candida albicans can activate the NF-κB inflammatory response through the Dectin-1→Syk→CARD9 signaling axis, while its regulatory factor Dal81 can inhibit the excessive activation of NF-κB by alkalizing the intracellular pH, forming a dynamic balance of “activation-inhibition” to achieve immune evasion [33].
 |
 |
|
(A) |
(B) |
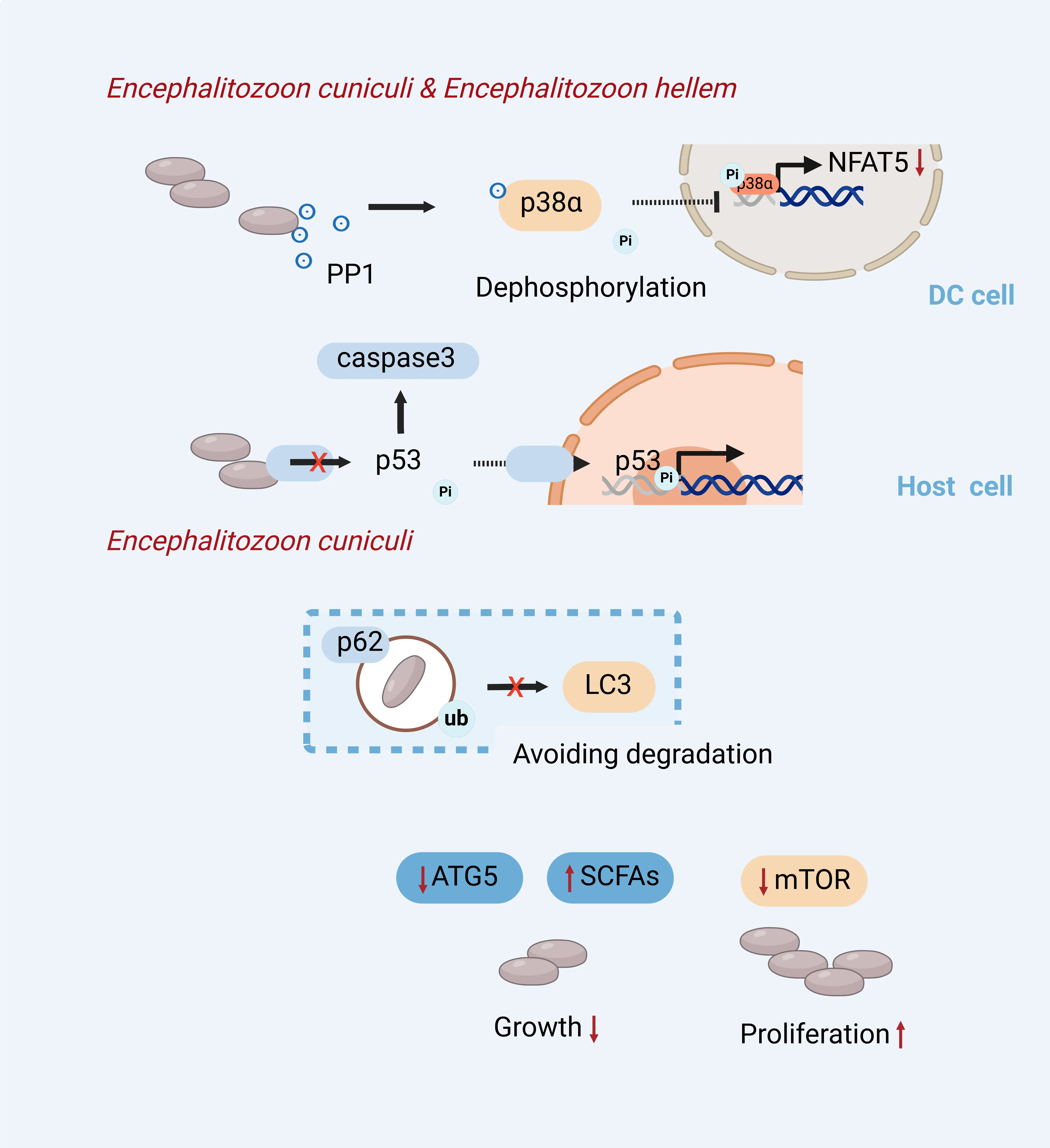
Figure 2. Fine-tuning of host signaling pathways by microsporidia and a representative fungal pathogen Candida albicans. (A) Candida albicans secretes candidin, which activates the p38 MAPK pathway in epithelial cells, inducing the release of pro-inflammatory factors, while simultaneously trans-activating the EGFR-ERK pathway to regulate cellular damage and repair; Candida albicans cell wall β-glucan activates NF-κB via the Dectin-1–Syk–CARD9 signaling axis to initiate an inflammatory response, while its regulatory factor Dal81 inhibits excessive NF-κB activation by alkalizing intracellular pH. (B) E. hellem/E. cuniculi actively secretes the serine/threonine protein phosphatase PP1, which inhibits the phosphorylation and activation of p38α in dendritic cells (DCs) through dephosphorylation, thereby blocking the signaling of downstream transcription factors such as NFAT5; Microsporidia of the genus Encephalitozoon effectively inhibit the cleavage and activation of host caspase-3 and block the phosphorylation and nuclear translocation of p53, thereby suppressing p53’s transcriptional regulatory function; In the early stages, E. cuniculi is marked by host ubiquitin and p62 to initiate heterologous autophagy, but it is able to evade degradation by autophagosomes by failing to recruit LC3, thereby hijacking the host autophagy pathway to obtain nutrients for itself. Inhibition of the upstream mTOR pathway activates autophagy and significantly promotes microsporidian proliferation, whereas knocking down the key autophagy gene ATG5 or inhibiting autophagy using short-chain fatty acids (SCFAs)—metabolites produced by the gut microbiota—significantly suppresses parasite growth.(p53:Key intracellular transcription factors;LC3:Autophagosome marker protein).
In contrast to the above typical fungal pathogens, the targeted signaling pathways of microsporidia in host cells have been revealed more recently. Furthermore, as the infection progresses and the individual’s life course unfolds, it regulates various signaling pathways. The TLR-Myd88/TRAF pathway is the first one, and the TLR-signaling pathways stand out in a single-cell analysis of microsporidia (E. cuniculi)- infected T cells. The up-regulation of TLR2 but down-regulation of effector cytokines indicates that TLR-Myd88/TRAF pathway may be targeted and modulated [34]. The next pathway to be regulated is the MAPK pathway, which is activated by inflammatory signals. zoonotic microsporidia E. hellem secretes serine/threonine protein phosphatase PP1, directly targeting the p38/MAPK signaling pathway in dendritic cells (DCs), inhibiting the activation of p38 MAPK protein, suppressing downstream transcriptional factors and genes expression. Consequently, markedly lower levels of pro-inflammatory cytokines such as IL-12, IL-6, and TNF-a are expressed, and the DCs function and maturation were impaired [15].
In the later stages of the infection, zoonotic microsporidia also ensure intracellular survival by modulating the p53-mediated apoptotic pathway [35]. Members of the genus Encephalitozoon effectively inhibit the cleavage and activation of host caspase-3, and block the phosphorylation and nuclear translocation of p53, thereby abolishing the transcriptional regulatory function of p53. This prevents apoptosis of infected cells and provides a stable proliferative niche for the continuous propagation of microsporidia.
Besides, the autophagic pathway and the mTOR pathway are also targets of zoonotic microsporidia. E. cuniculi achieves efficient nutrient acquisition and proliferation by hijacking the host autophagic pathway by interfering with LC3 recruitment. As a result, E. cuniculi can escape degradation in autolysosomes and even supply nutrients for proliferation [25].
4. Immune Regulation
Signaling fine-tuning often serves as the upstream trigger for downstream immune modulation and therefore represents a critical battleground for intracellular pathogens. In the following section, we detail how zoonotic microsporidia reshape host immunity—from pattern recognition to T cell responses.
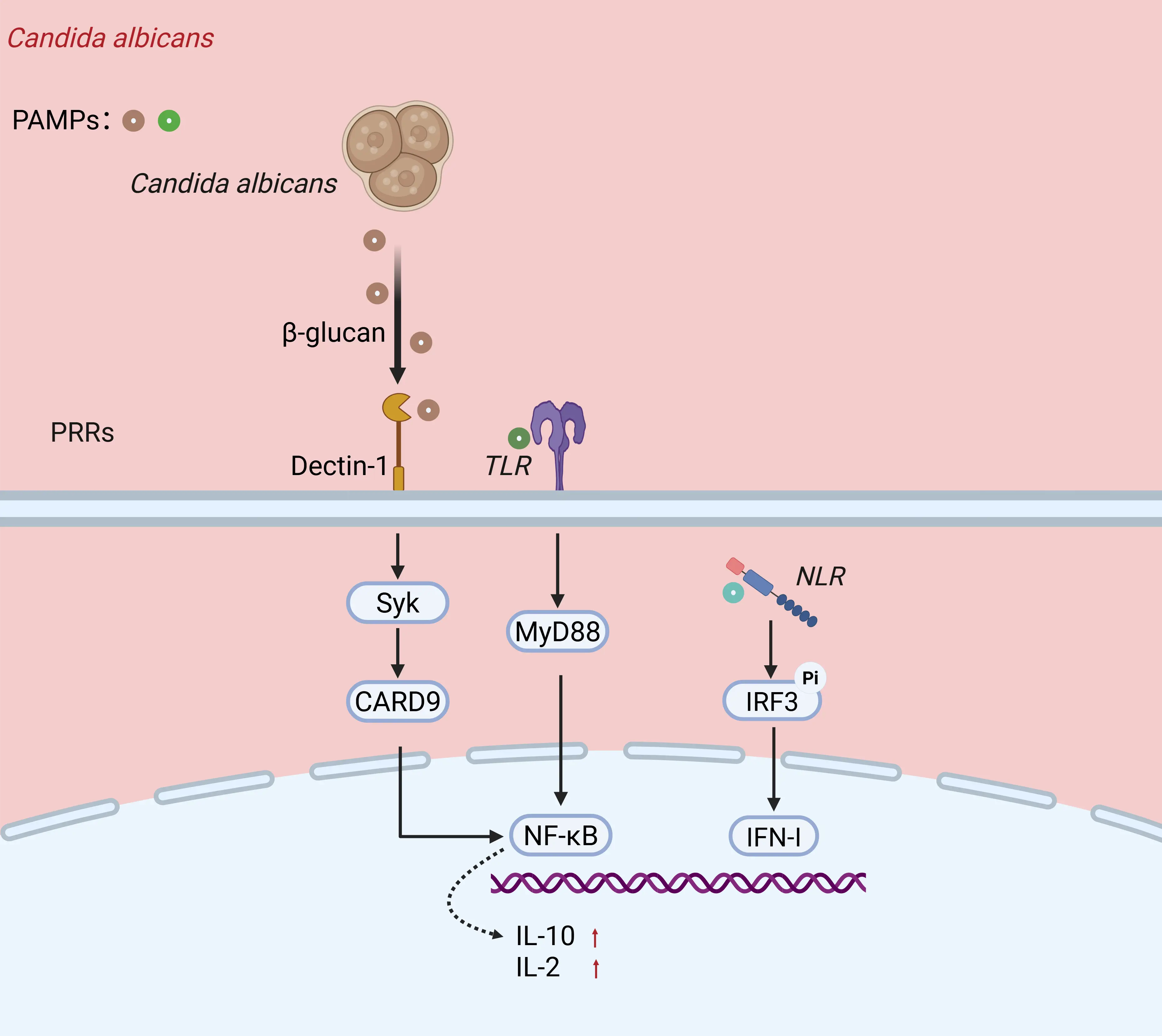
Both Candida albicans and Cryptococcus neoformans are facultative parasitic fungi capable of targeting host innate and adaptive immune pathways through cell wall components, secreted effector molecules, and toxins (Figure 3A). By modulating immune activation and evasion, they achieve a dynamic balance that facilitates host invasion and colonization. Candida albicans relies on pattern recognition receptors (PRRs), mainly Toll-like receptors (TLRs), C-type lectin receptors (CLRs), and NOD-like receptors (NLRs) [36]. Dectin-1 specifically recognizes β-glucan on the cell wall of Candida albicans, and upon activation initiates a series of host cellular immune responses, while acting synergistically with TLRs to regulate host antifungal immunity [37].
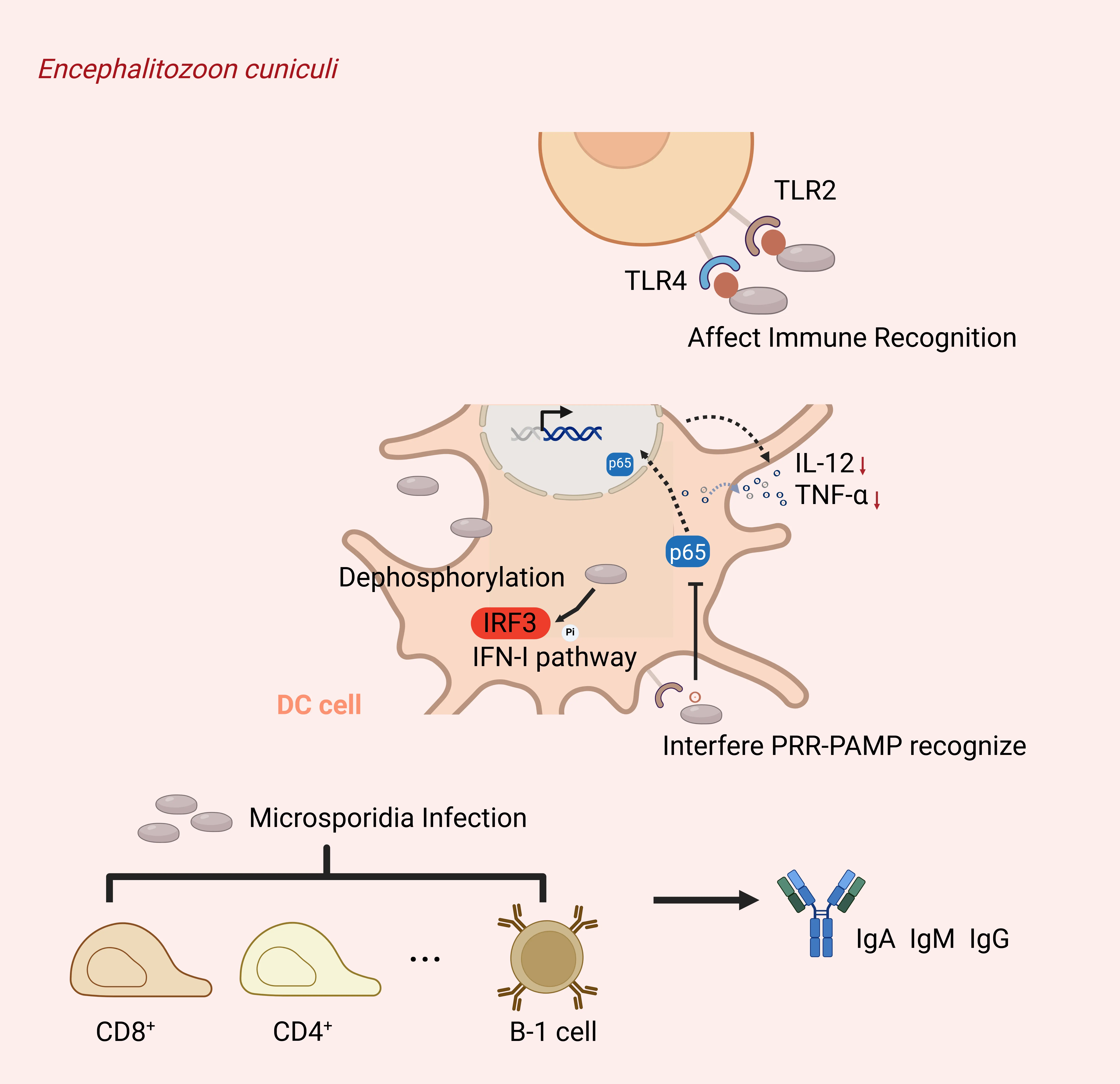
As the obligate intracellular parasitic fungi with highly reduced genomes and lacking typical immunogenic fungal PAMPs, microsporidia usually do not stimulate intense inflammatory responses during invasion [29,38]. Tthe host immune responses are often suppressed during microsporidia infection alongside the maintenance of host cell survival, which is closely aligned with their intracellular parasitic requirements (Figure 3B). Innate immune suppression by microsporidia is more frequently reported. For instance, E. hellem targets p38 MAPK to inhibit the DCs phagocytosis, cell maturation, and antigen presentation [15]. Microsporidia can employ effector proteins to disrupt the PRR–PAMP recognition process, inhibit nuclear translocation of the NF-κB p65 subunit, and reduce the release of pro-inflammatory factors. IRF3 phosphorylation and the activation of the type I interferon (IFN-I) signaling pathway are also suppressed. These mechanisms prevent excessive innate immune responses and create a stable intracellular environment conducive to microsporidian proliferation [39].
 |
 |
|
(A) |
(B) |
Figure 3. Immune regulations by microsporidia and a representative fungal pathogen. (A) Candida albicans is recognized by Dectin-1, Toll-like receptors (TLRs), and NOD-like receptors (NLRs) on the surface of macrophages through its pathogen-associated signaling molecules (such as β-glucan), thereby activating downstream signaling molecules including Syk, MyD88, and CARD9, which ultimately leads to upregulation of IL-2 and IL-10 expression. (B) Microsporidia E. cuniculi interferes with the TLR2/TLR4-mediated recognition by host dendritic cells, disrupting the PRR-PAMP recognition process. This leads to the dephosphorylation of p65 and the inhibition of the IRF3 and IFN-I pathways, thereby reducing the secretion of IL-12 and TNF-α, and ultimately attenuating the immune response of CD4⁺, CD8⁺ T cells, and B-1 cells. (Dectin-1: Specifically recognizes β-glucan on the cell wall of Candida albicans PRR-PAMP: The process by which microsporidia recognize pathogen-associated molecular patterns via host pattern recognition receptors.
The host adaptive immune system could also be targeted for regulation by zoonotic microsporidia. It’s reported that microsporidia E. cuniculi stimulate cellular immunity, especially on the perforin and granzymes producing CD8⁺ cytotoxic T lymphocytes. In addition, other immune cells such as CD4⁺ T cells, γδ T cells, and B-1 cells are also activated to participate in immune regulation [34]. In addition, microsporidia stimulate the host to produce antibodies, including IgG and IgM, to facilitate neutralization and phagocytosis [40]. Moreover, microsporidia are reported to establish a favorable parasitic immune environment by inhibiting host cell apoptosis by downregulating the expression of pro-apoptotic proteins or inhibiting the cleavage of pro-caspases [35].
5. Epigenetic Modification
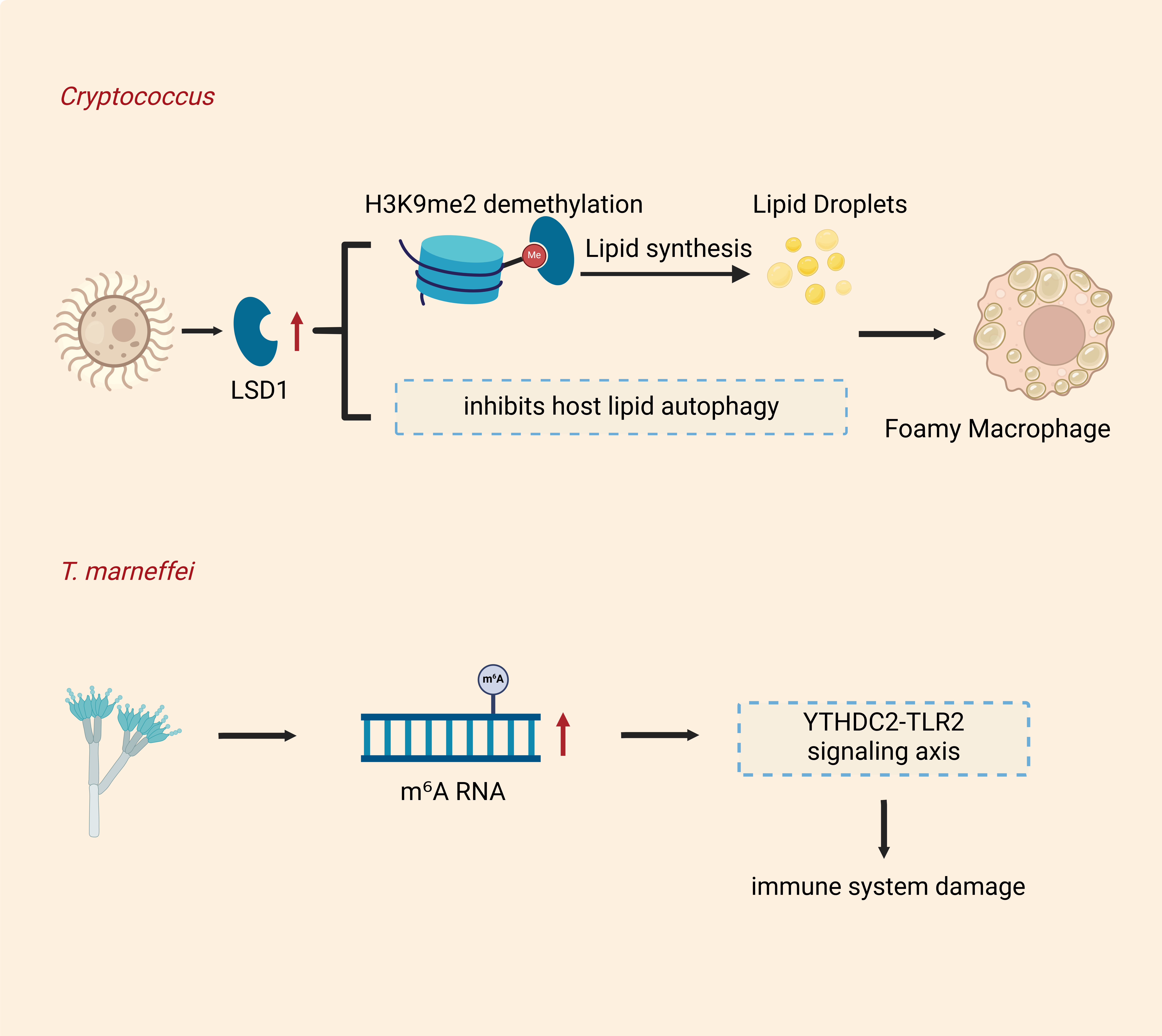
Beyond immediate immune control, recent evidence reveals that microsporidia also impose long‑lasting changes on host cells through epigenetic reprogramming. Epigenetics, as a heritable form of gene expression regulation independent of DNA sequence changes, serves as a key battleground for pathogens to regulate their hosts and plays a significant role in the proliferation and spread of fungal pathogens following invasion. As shown (Figure 4A), when Cryptococcus neoformans infects hosts, it activates and upregulates the expression of the histone demethylase LSD1, LSD1 selectively binds to the promoters of genes associated with lipid uptake (Ldlr, Cd36) and biosynthesis (Plin2), reducing the levels of the repressive histone mark H3K9me2 and thereby activating the transcription of these genes. Concurrently, it directly inhibits host lipid autophagy (by blocking the lysosomal degradation of lipid droplets), which ultimately leads to abnormal lipid accumulation and the formation of foam macrophages [20]. Talaromyces marneffei primarily infects immunocompromised individuals and is an important opportunistic pathogen. T. marneffei infection induces elevated levels of m6A RNA modification, as well as IncRNAs in host cells. These modifications alter the characteristics of the host transcriptome, exacerbate damage to the host immune system via the YTHDC2-TLR2 signaling axis, and simultaneously increase the susceptibility of host cells to HIV, accelerating disease progression in co-infected patients [41,42].
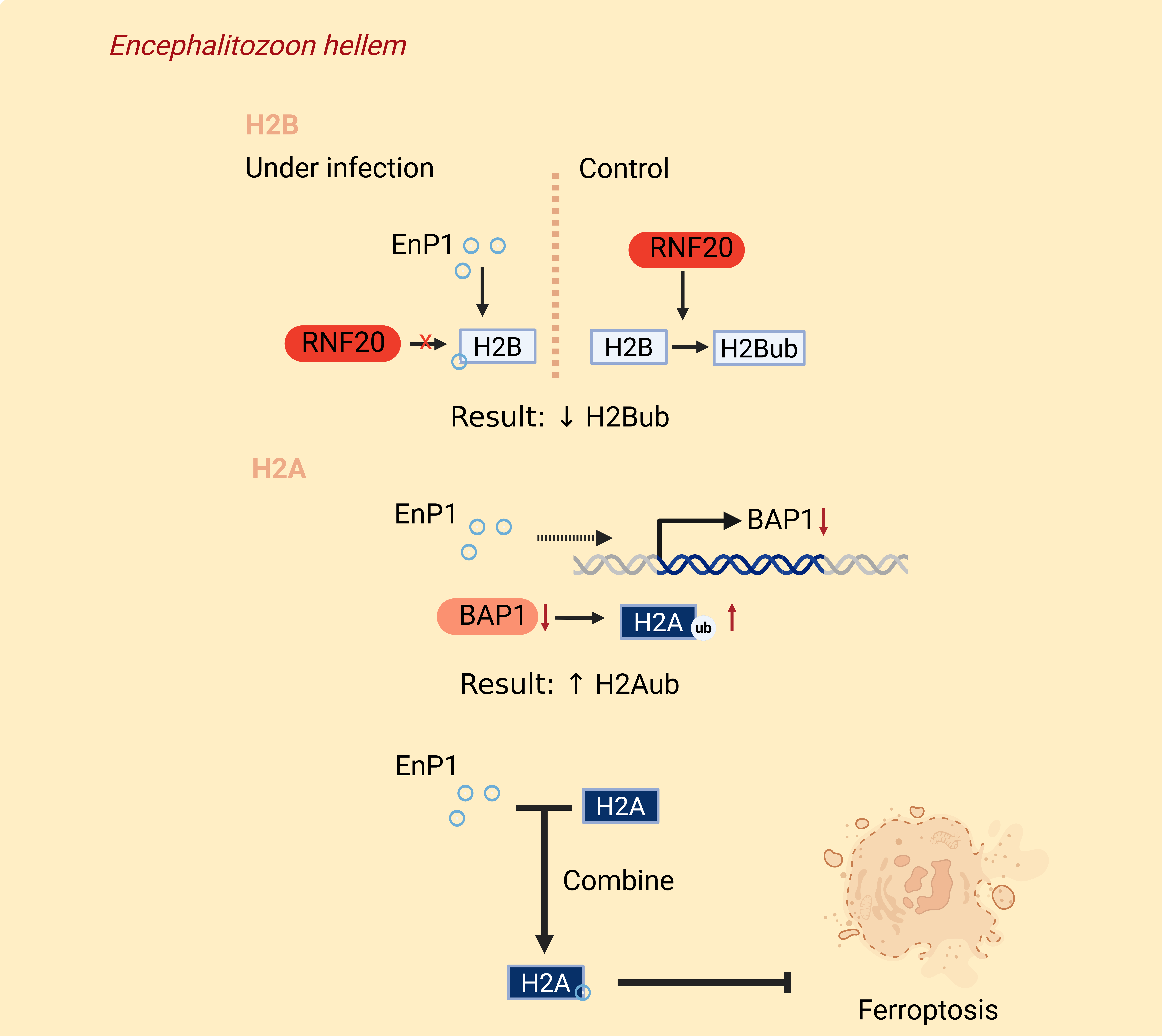
Like T. marneffei, zoonotic microsporidia primarily infect immunocompromised individuals and are capable of modulating host epigenetic regulation. However, the mechanisms differ (Figure 4B): T. marneffei infection affects RNA modifications in host cells, whereas the microsporidian E. hellem regulates host histone ubiquitination. Microsporidian EnP1 is a nucleus-targeted effector that can modify the host cell environment. This effector contains a functional signal peptide (SP) and two NLSs, highly conserved within the genus Microsporidium. Following infection, EnP1 can enter the host cell nucleus, whereas the double NLS-deleted mutant (EnP1ΔNLS) localizes exclusively to the cytoplasm and fails to translocate into the nucleus. EnP1 does not directly bind RNF20, the key ubiquitin ligase responsible for H2Bub; instead, it competitively binds to H2B, thereby blocking the interaction between RNF20 and H2B and inhibiting H2Bub. Furthermore, knocking down RNF20 or overexpressing H2BK120R (which cannot be ubiquitinated) mimics the effects of EnP1, significantly promoting microsporidian proliferation [39].
 |
 |
|
(A) |
(B) |
Figure 4. Epigenetic modification by microsporidia and other fungal pathogens. (A) When Cryptococcus neoformans infects a host, it activates and upregulates LSD1 expression. LSD1 selectively binds to the promoters of genes associated with lipid uptake (Ldlr, Cd36) and biosynthesis (Plin2), and reduces the levels of the repressive histone mark H3K9me2, thereby activating the transcription of these genes. Concurrently, it directly inhibits host lipid autophagy, ultimately leading to abnormal lipid accumulation and the formation of foam macrophages. Infection with Talaromyces marneffei induces elevated levels of m⁶A RNA modifications in host cells. This abnormal modification alters the characteristics of the host transcriptome and exacerbates damage to the host immune system via the YTHDC2-TLR2 signaling axis. (B) Following infection by E. hellem, EnP1 is secreted into the host cell nucleus, where it competitively binds to H2B, thereby blocking the interaction between RNF20 and H2B and consequently inhibiting H2Bub; Overexpression of EnP1 or microsporidian infection significantly downregulates only the mRNA and protein expression of the deubiquitinating enzyme BAP1, and since EnP1 does not directly interact with BAP1, this suggests that EnP1 elevates H2A monoubiquitin (H2Aub) levels by inhibiting BAP1 expression. (LSD1: histone demethylase; EnP1: microsporidian capsid protein, which is also a nuclear-targeted effector protein secreted by microsporidia; BAP1: deubiquitinating enzyme, the core catalytic subunit of the PR-DUB complex).
At the same time, EnP1 also binds directly to histone H2A, and together inhibit host ferroptosis, although the mechanisms are slightly different: H2A monoubiquitination (H2Aub) is mediated by RING1A/B of the PRC1 complex, whereas deubiquitination is driven by BAP1 of the PR-DUB complex. This experimental evidence has also confirmed that EnP1 overexpression or microsporidia infection did not affect the transcription or protein levels of RING1A/B or USP16 (another H2A deubiquitinase), but significantly downregulated the mRNA and protein expression of BAP1. EnP1 does not directly interact with BAP1, indicating that EnP1 elevates H2A monoubiquitin (H2Aub) levels by suppressing BAP1 expression [17,18]. These mechanistically distinct yet functionally synergistic strategies of host epigenetic regulation employed by microsporidia and other fungal pathogens illustrate the evolutionary sophistication of microsporidia within the fungal kingdom.
Collectively, these four dimensions—metabolic, signaling, immune, and epigenetic—operate as an integrated network rather than isolated strategies.
6. Conclusions
Zoonotic microsporidia, as obligate intracellular fungal pathogens, have evolved a sophisticated, multi-layered regulatory network to reprogram host cell functions for their survival, proliferation, and immune evasion. This review systematically dissects these regulatory strategies from four interconnected dimensions: metabolic reprogramming, signaling pathway modulation, immune regulation, and epigenetic remodeling.
At the metabolic level, microsporidia exploit host ATP, amino acids, and nucleotides via mitochondrion–parasitophorous vacuole contact sites (e.g., EhSSP1-VDAC interaction), hijack the ERAD pathway (e.g., EhPTP4), and subvert host autophagy to acquire biosynthetic building blocks—contrasting sharply with canonical fungal pathogens that often suppress autophagy. At the signaling level, secreted effectors such as PP1 directly dampen p38 MAPK activation in dendritic cells, while manipulation of TLR-MyD88, p53-mediated apoptosis, and mTOR-autophagy axes ensures a non-lytic, replication-permissive intracellular niche. Immunologically, microsporidia actively suppress both innate (e.g., inhibition of DC maturation, NF-κB, and type I IFN) and adaptive (e.g., modulation of CD8⁺ T cell cytotoxicity and antibody responses) immunity, establishing a finely tuned balance between host tolerance and parasite persistence. Epigenetically, the nuclear effector EnP1 exemplifies a novel mechanism by competitively blocking H2B monoubiquitination and downregulating BAP1 to elevate H2A ubiquitination, thereby inhibiting host ferroptosis and remodeling transcriptional programs favorable to parasite proliferation—a strategy distinct from the RNA m⁶A modification employed by other opportunistic fungi like Talaromyces marneffei.
Despite these advances, several critical gaps remain. First, the full repertoire of secreted effectors and their host targets is far from complete, particularly for Enterocytozoon bieneusi, which lacks robust in vitro culture systems. Second, the crosstalk among metabolic, signaling, immune, and epigenetic layers—e.g., how EnP1-mediated histone modification influences metabolic gene expression—remains poorly understood. Third, most studies rely on Encephalitozoon models; comparative analyses across different zoonotic genera and host species are urgently needed to delineate conserved versus species-specific regulatory principles.
Looking forward, integrating high-resolution structural biology, single-cell multi-omics, and CRISPR-based host–pathogen interaction screens will accelerate the discovery of druggable host targets and parasite-derived virulence factors. For example, cryo-electron microscopy (Cryo-EM) and cryo-electron tomography (Cryo-ET) can be used to elucidate the substrate selectivity and transport cycle of microsporidian ATP transporters, thereby guiding the design of competitive inhibitors to block the theft of host energy by microsporidians. Single-cell omics technologies can address key challenges in microsporidia research: the spatiotemporal heterogeneity of the infection stage and the heterogeneity of the host immune response. As for CRISPR technology, it can be used to screen for genes essential for microsporidia to infect their hosts and to identify host immune pathways targeted by microsporidia effector proteins. Given that microsporidiosis primarily threatens immunocompromised populations and agricultural animals, deciphering these regulatory networks holds translational promise: host-directed therapies (e.g., modulating p38 MAPK or ferroptosis pathways) and epigenetic inhibitors could complement conventional antiparasitic drugs. Moreover, as a minimalist model of intracellular parasitism within the fungal kingdom, zoonotic microsporidia offer unique insights into the evolution of host manipulation—from metabolic dependence to epigenetic coercion—illuminating fundamental principles that may apply to other intracellular pathogens.
In conclusion, host regulatory mechanisms of zoonotic microsporidia constitute a complex, highly specialized, and evolutionarily convergent network. Unraveling this network not only deepens our understanding of microsporidia pathogenesis but also establishes a conceptual and technological foundation for next-generation interventions, ultimately strengthening public health defenses and sustaining animal husbandry against these emerging pathogens.
Author Contributions
Writing—Original Draft Preparation, L.L-T., J.J., M.Z.; Visualization, J.J., M.Z.; Writing—Review & Editing, L.L-T., Q.H., M.R., J.C., J.B.; Supervision, Z.Z., J.B.; Project Administration, J.B.; Funding Acquisition, J.B.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Funding
This study was supported by The National Natural Science Foundation of China (No. 32570231); National Undergraduate Innovation and Entrepreneurship Training Program Project (No. 202410635032).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Hirt RP, Logsdon JM, Healy B, Dorey MW, Doolittle WF, Embley TM. Microsporidia are related to Fungi: Evidence from the largest subunit of RNA polymerase II and other proteins. Proc. Nat. Acad. Sci. USA 1999, 96, 580–585. DOI:10.1073/pnas.96.2.580 [Google Scholar]
- Han B, Weiss LM. Microsporidia: Obligate Intracellular Pathogens Within the Fungal Kingdom. Microbiol. Spectrum 2017, 5. DOI:10.1128/microbiolspec.FUNK-0018-2016 [Google Scholar]
- Mathis A, Weber R, Deplazes P. Zoonotic Potential of the Microsporidia. Clin. Microbiol. Rev. 2005, 18, 423–445. DOI:10.1128/CMR.18.3.423-445.2005 [Google Scholar]
- Prasertbun R, Mori H, Pintong AR, Sanyanusin S, Popruk S, Komalamisra C, et al. Zoonotic potential of Enterocytozoon genotypes in humans and pigs in Thailand. Vet. Parasitol. 2017, 233, 73–79. DOI:10.1016/j.vetpar.2016.12.002 [Google Scholar]
- Weber R, Kuster H, Visvesvara GS, Bryan RT, Schwartz DA, Luthy R. Disseminated Microsporidiosis Due to Encephalitozoon hellem: Pulmonary Colonization, Microhematuria, and Mild Conjunctivitis in a Patient with AIDS. Clin. Infect. Dis. 1993, 17, 415–419. DOI:10.1093/clinids/17.3.415 [Google Scholar]
- Nourrisson C, Hamane S, Bonhomme J, Durieux MF, Foulquier JB, Lesthelle S, et al. Case series of intestinal microsporidiosis in non-HIV patients caused by Encephalitozoon hellem. Emerg. Microbes Infec. 2023, 12, 2258997. DOI:10.1080/22221751.2023.2258997 [Google Scholar]
- Vávra J, Lukeš J. Microsporidia and ‘The Art of Living Together’. Adv. Parasitol. 2013, 253–319. DOI:10.1016/B978-0-12-407706-5.00004-6 [Google Scholar]
- Huang Q, Chen J, Lv Q, Long M, Pan G, Zhou Z. Germination of Microsporidian Spores: The Known and Unknown. J. Fungi 2023, 9, 774. DOI:10.3390/jof9070774 [Google Scholar]
- Meng XZ, Luo B, Tang XY, He Q, Xiong TR, Fang ZY, et al. Pathological analysis of silkworm infected by two microsporidia Nosema bombycis CQ1 and Vairimorpha necatrix BM. J. Inverteb. Pathol. 2018, 153, 75–84. DOI:10.1016/j.jip.2017.12.005 [Google Scholar]
- Pan G, Bao J, Ma Z, Song Y, Han B, Ran M, et al. Invertebrate host responses to microsporidia infections. Dev. Comp. Immunol. 2018, 83, 104–113. DOI:10.1016/j.dci.2018.02.004 [Google Scholar]
- Flynn PM. Emerging Diarrheal Pathogens: Cryptosporidium parvum, Isospora belli, Cyclospora Species, and Microsporidia. Pediatr. Ann. 1996, 25, 480–487. DOI:10.3928/0090-4481-19960901-04 [Google Scholar]
- Mota P, Rauch CA, Edberg SC. Microsporidia andCyclospora: Epidemiology and Assessment of Risk from the Environment. Crit. Rev. Microbiol. 2000, 26, 69–90. DOI:10.1080/10408410091154192 [Google Scholar]
- Tang L, Sabi MM, Fu M, Guan J, Wang Y, Xia T, et al. Host cell manipulation by microsporidia secreted effectors: Insights into intracellular pathogenesis. J. Eukaryotic Microbiol. 2024, 71, e13029. DOI:10.1111/jeu.13029 [Google Scholar]
- Han B, Ma Y, Tu V, Tomita T, Mayoral J, Williams T, et al. Microsporidia Interact with Host Cell Mitochondria via Voltage-Dependent Anion Channels Using Sporoplasm Surface Protein 1. mBio 2019, 10. DOI:10.1128/mBio.01944-19 [Google Scholar]
- Bao J, Tang Y, Chen Y, Jin J, Wang X, An G, et al. E. hellemSer/Thr protein phosphatase PP1 targets the DC MAPK pathway and impairs immune functions. Life Sci. Alliance 2024, 7, e202302375. DOI:10.26508/lsa.202302375 [Google Scholar]
- Ran M, Bao J, Li B, Shi Y, Yang W, Meng X, et al. Microsporidian Nosema bombycis secretes serine protease inhibitor to suppress host cell apoptosis via Caspase BmICE. PLoS Pathog. 2025, 21, e1012373. DOI:10.1371/journal.ppat.1012373 [Google Scholar]
- Liu F, Meng X, Pan G, Bao J, Chen J. The survival and escape strategies of microsporidia. Trend. Parasitol. 2026, 42, 249–263. DOI:10.1016/j.pt.2026.02.008 [Google Scholar]
- Guan J, Wang Y, Fu M, Tang L, Sabi MM, Zhu H, et al. EnP1 exploits H2Aub-dependent epigenetic reprogramming to promote microsporidia proliferation in host cells. PLoS Pathog. 2026, 22, e1013853. DOI:10.1371/journal.ppat.1013853 [Google Scholar]
- Pellon A, Sadeghi Nasab SD, Bidkhori G, Griffiths JS, Vaga S, Begum N, et al. Fungal infection drives metabolic reprogramming in epithelial cells via aerobic glycolysis and an alternative TCA cycle shunt. Sci. Adv. 2026, 12, eaea0405. DOI:10.1126/sciadv.aea0405 [Google Scholar]
- Lohia GK, Shah A, Balaji KN. Histone demethylase LSD1 regulates lipid homeostasis during Cryptococcus neoformans infection. iScience 2025, 28, 113405. DOI:10.1016/j.isci.2025.113405 [Google Scholar]
- Hacker C, Sendra K, Keisham P, Filipescu T, Lucocq J, Salimi F, et al. Biogenesis, inheritance, and 3D ultrastructure of the microsporidian mitosome. Life Sci. Alliance 2024, 7, e202201635. DOI:10.26508/lsa.202201635 [Google Scholar]
- Watson AK, Williams TA, Williams BAP, Moore KA, Hirt RP, Embley TM. Transcriptomic profiling of host-parasite interactions in the microsporidian Trachipleistophora hominis. BMC Genom. 2015, 16, 983. DOI:10.1186/s12864-015-1989-z [Google Scholar]
- Hacker C, Howell M, Bhella D, Lucocq J. Strategies for maximizing ATP supply in the microsporidianEncephalitozoon cuniculi: direct binding of mitochondria to the parasitophorous vacuole and clustering of the mitochondrial porin VDAC. Cell. Microbiol. 2014, 16, 565–579. DOI:10.1111/cmi.12240 [Google Scholar]
- Luo J, He Q, Xu JZ, Xu C, Han YZ, Gao HL, et al. Microsporidia infection upregulates host energy metabolism but maintains ATP homeostasis. J. Inverteb. Pathol. 2021, 186, 107596. DOI:10.1016/j.jip.2021.107596 [Google Scholar]
- Panek J, Carriere E, Saleh MB, Sendra K, Kosta G, Korolchuk VI, et al. Microsporidian obligate intracellular parasites subvert autophagy of infected mammalian cells to promote their own growth. mBio 2025, 16, e01049-25. DOI:10.1128/mbio.01049-25 [Google Scholar]
- Rost‐Roszkowska MM, Poprawa I, Kaczmarek Ł. Autophagy as the cell survival in response to a microsporidian infection of the midgut epithelium of Isohypsibius granulifer granulifer (Eutardigrada: Hypsibiidae). Acta Zool. 2013, 94, 273–279. DOI:10.1111/j.1463-6395.2011.00552.x [Google Scholar]
- Han B, Polonais V, Sugi T, Yakubu R, Takvorian PM, Cali A, et al. The role of microsporidian polar tube protein 4 (PTP4) in host cell infection. PLoS Pathog. 2017, 13, e1006341. DOI:10.1371/journal.ppat.1006341 [Google Scholar]
- Bao J, Liu L, An Y, Ran M, Ni W, Chen J, et al. Nosema bombycis suppresses host hemolymph melanization through secreted serpin 6 inhibiting the prophenoloxidase activation cascade. J. Inverteb. Pathol. 2019, 168, 107260. DOI:10.1016/j.jip.2019.107260 [Google Scholar]
- Corradi N, Slamovits CH. The intriguing nature of microsporidian genomes. Brief. Funct. Genom. 2011, 10, 115–124. DOI:10.1093/bfgp/elq032 [Google Scholar]
- Prybylski N, Fayet M, Dubuffet A, Delbac F, Kocer A, Gardarin C, et al. Ricin B lectin-like proteins of the microsporidian Encephalitozoon cuniculi and Anncaliia algerae are involved in host-cell invasion. Parasitol. Int. 2022, 87, 102518. DOI:10.1016/j.parint.2021.102518 [Google Scholar]
- Moyes DL, Wilson D, Richardson JP, Mogavero S, Tang SX, Wernecke J, et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 2016, 532, 64–68. DOI:10.1038/nature17625 [Google Scholar]
- Nikou SA, Zhou C, Griffiths JS, Kotowicz NK, Coleman BM, Green MJ, et al. The Candida albicans toxin candidalysin mediates distinct epithelial inflammatory responses through p38 and EGFR-ERK pathways. Sci. Signal. 2022, 15, eabj6915. DOI:10.1126/scisignal.abj6915 [Google Scholar]
- Huang X, Chen G, Wu L, Zou Y, Zhang L, Li S, et al. Coordinated regulation of pH alkalinization by two transcription factors promotes fungal commensalism and pathogenicity. Nat. Commun. 2025, 16, 7855. DOI:10.1038/s41467-025-62953-x [Google Scholar]
- Tang Y, Cao L, Jin J, Li T, Chen Y, Lu Y, et al. Single-cell transcriptional responses of T cells during microsporidia infection. Commun. Biol. 2025, 8, 567. DOI:10.1038/s42003-025-07990-4 [Google Scholar]
- del Aguila C, Izquierdo F, Granja AG, Hurtado C, Fenoy S, Fresno M, et al. Encephalitozoon microsporidia modulates p53-mediated apoptosis in infected cells. Int. J. Parasitol. 2006, 36, 869–876. DOI:10.1016/j.ijpara.2006.04.002 [Google Scholar]
- Netea MG, Brown GD, Kullberg BJ, Gow NAR. An integrated model of the recognition of Candida albicans by the innate immune system. Nat. Rev. Microbiol. 2008, 6, 67–78. DOI:10.1038/nrmicro1815 [Google Scholar]
- Wang Y. Looking intoCandida albicansinfection, host response, and antifungal strategies. Virulence 2015, 6, 307-308. DOI:10.1080/21505594.2014.1000752 [Google Scholar]
- Katinka MD, Duprat S, Cornillot E, Méténier G, Thomarat F, Prensier G, et al. Genome sequence and gene compaction of the eukaryote parasite Encephalitozoon cuniculi. Nature 2001, 414, 450–453. DOI:10.1038/35106579 [Google Scholar]
- Guan J, Tang L, Wang Y, Fu M, Xia T, Zheng K, et al. Microsporidian EnP1 alters host cell H2B monoubiquitination and prevents ferroptosis facilitating microsporidia survival. Proc. Nat. Acad. Sci. USA 2024, 121, e2400657121. DOI:10.1073/pnas.2400657121 [Google Scholar]
- Han B, Pan G, Weiss LM. Microsporidiosis in Humans. Clin. Microbiol. Rev. 2021, 34, e00010-20. DOI:10.1128/CMR.00010-20 [Google Scholar]
- Chu J, Zheng R, Chen H, Chen Y, Lin Y, Li J, et al. Dynamic m6A profiles reveal the role of YTHDC2‐TLR2 signaling axis in Talaromyces marneffei infection. J. Med. Virol. 2024, 96, e29466. DOI:10.1002/jmv.29466 [Google Scholar]
- Li Y, Chen H, Li S, Li Y, Liu G, Bai J, et al. LncSSBP1 Functions as a Negative Regulator of IL-6 Through Interaction With hnRNPK in Bronchial Epithelial Cells Infected With Talaromyces marneffei. Front. Immunol. 2020, 10. DOI:10.3389/fimmu.2019.02977 [Google Scholar]