Kv1.5 Inhibition in Atrial Fibrillation: Molecular Mechanisms, Translational Challenges, and Implications for Equitable Rhythm Control

Kv1.5 Inhibition in Atrial Fibrillation: Molecular Mechanisms, Translational Challenges, and Implications for Equitable Rhythm Control

Received: 30 March 2026 Revised: 10 April 2026 Accepted: 15 May 2026 Published: 28 May 2026

© 2026 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
1. Introduction
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia, affecting an estimated 33.5 million individuals worldwide in 2010 [1], with more recent data indicating this number has risen to nearly 59 million by 2019 [2]. The lifetime risk of developing AF approaches 1 in 3 adults, and prevalence continues to rise with population aging and increasing cardiometabolic disease burden [3,4]. Despite decades of therapeutic development, pharmacologic rhythm control remains limited by the toxicity and ventricular proarrhythmia risk of available antiarrhythmic agents, underscoring the need for safer, more targeted treatment strategies.
AF is often asymptomatic early, which contributes to underdiagnosis, particularly in socioeconomically disadvantaged populations [5]. Yet its consequences are severe: AF increases the risk of stroke three- to fivefold compared with individuals without AF and contributes to heart failure (HF) exacerbation, reduced quality of life, and increased all-cause mortality [6].
1.1. Disparities in Detection and Outcomes
Despite its prevalence and severity, AF burden is not equitably distributed: many racial and ethnic minority groups have lower rates of clinically diagnosed AF compared with White patients, a pattern likely driven by differences in healthcare access, symptom recognition, and detection, rather than true differences in AF prevalence [7].
Comorbidities such as CKD and heart failure further complicate rhythm-control therapy, as agents such as sotalol and dofetilide require renal dose adjustment and close ECG monitoring, limiting their use in medically complex patients and low-resource settings [8,9].
1.2. Economic and Public Health Impact
The economic burden of AF is substantial, with more than 750,000 hospitalizations annually in the US alone [10] and estimated incremental healthcare costs of $6–26 billion per year [11,12]. Globally, AF affects more than 52 million people, and the absolute burden has more than doubled since 1990, disproportionately affecting low- and middle-income regions where access to rhythm-control therapies remains limited [13,14,15]. The need for intensive monitoring during the initiation of antiarrhythmic therapies further increases healthcare utilization and exacerbates disparities in access to care, highlighting the urgent need for safer, more accessible rhythm-control options [11,12].
1.3. AF Epidemiology and the Racial Paradox
A well-described epidemiologic paradox exists in atrial fibrillation (AF): despite a higher burden of traditional cardiovascular risk factors, Black individuals consistently demonstrate a lower prevalence and incidence of AF compared with White individuals [16,17]. This phenomenon, often termed the “atrial fibrillation racial paradox”, has been observed across multiple epidemiologic studies and population-based cohorts in the United States. For example, large cohort studies including the ARIC and REGARDS investigations have demonstrated that Black participants have substantially lower AF prevalence despite higher rates of hypertension, diabetes, obesity, and other cardiometabolic risk factors [18,19,20]. Although the mechanisms underlying this paradox remain incompletely understood, proposed explanations include genetic factors, including variants in ion channel genes such as KCNA5, differences in atrial structure or electrophysiology, and potential differences in detection or clinical ascertainment [16].
Despite the lower prevalence of AF, Black individuals who develop the arrhythmia experience a higher burden of cardiovascular complications, including stroke and mortality [16,18,19]. Differences in treatment patterns may contribute to these outcome disparities, as several studies have shown that Black patients are less likely to receive rhythm-control interventions such as cardioversion, catheter ablation, or specialist electrophysiology care [18]. Additionally, prior analyses have demonstrated disparities in anticoagulation therapy and anticoagulation control, which may further increase the risk of thromboembolic complications. Together, these differences in treatment access and management patterns may partially explain the disproportionate burden of AF-related complications observed in Black populations [18].
1.4. Detection and Diagnostic Bias
Differences in AF detection and clinical recognition likely contribute to the observed epidemiologic patterns. Studies, such as the Multi-Ethnic Study of Atherosclerosis (MESA) investigation, have shown that clinically detected AF is significantly less common in Black individuals, whereas long-term ambulatory monitoring identifies comparable rates of subclinical arrhythmia across racial groups [18,19]. Because AF is frequently asymptomatic or paroxysmal in these populations, disparities in access to continuous monitoring or specialty cardiovascular care, often driven by structural social determinants of health, influence the likelihood of a timely diagnosis [21]. These diagnostic and structural factors, combined with differences in underlying atrial disease and cardiovascular risk profiles, ultimately shape the observed disparities in AF prevalence and clinical outcomes [18,21].
Chronic kidney disease (CKD) and AF share a bidirectional relationship. AF prevalence rises markedly with declining renal function, reaching 15–20% in advanced CKD and end-stage renal disease populations [22], and incident AF occurs two- to three-fold more frequently in ESRD than in the general population [23]. Conversely, AF has been identified as an independent risk factor for developing ESRD among patients with underlying CKD [8]. Declining renal function is independently associated with higher thromboembolic risk in patients with atrial fibrillation, and incorporation of eGFR < 60 mL/min into the R2CHADS2 score has been shown in validation cohorts to mildly improve stroke risk prediction, although current clinical guidelines continue to rely primarily on CHA2DS2-VASc for routine risk stratification [24,25].
Renal impairment further complicates rhythm-control therapy. Dofetilide and sotalol are predominantly renally cleared (~80% unchanged), requiring dose adjustment with declining creatinine clearance; dofetilide is contraindicated when CrCl < 20 mL/min [26], and sotalol must be reduced below 60 mL/min, narrowing options for advanced CKD patients [27]. Clinically, individuals with combined AF and CKD experience substantially higher mortality and hospitalization rates, with particularly poor outcomes when heart failure coexists [28,29], underscoring the vulnerability of this cardiorenal population and the necessity of accurate renal assessment to avoid diagnostic and therapeutic inequities.
Emerging genetic evidence suggests that variation in KCNA5, the gene encoding Kv1.5, may contribute to differences in AF susceptibility across populations. Both loss-of-function and gain-of-function variants in KCNA5 have been identified in individuals with early-onset lone AF, with loss-of-function mutations eliminating IKur current entirely and gain-of-function mutations altering atrial action potential duration through changes in Ca2+ homeostasis [30]. Notably, these variants have been reported across Danish, European, and Asian populations, suggesting that KCNA5 polymorphisms are not population-specific but may differ in frequency and functional consequence across ethnic groups [30]. This is particularly relevant given the broader observation that cardiac potassium channel variants occur at a higher frequency in Black individuals compared with White individuals across several ion channel genes, pointing to meaningful differences in the genetic architecture of cardiac repolarization across populations [31]. Together, these findings suggest that genetic variation in Kv1.5 may contribute to inter-individual differences in atrial electrophysiology and could become relevant to precision pharmacology. However, direct evidence linking KCNA5 variation to population-level differences in AF presentation or response to Kv1.5-targeted therapy is limited. At present, direct population-comparative data on cardiac Kvβ1.2 expression or function are lacking, showing an important knowledge gap for future precision-targeted studies of Kv1.5-directed therapy.
These limitations highlight an important gap in current antiarrhythmic therapy. Many rhythm-control drugs require intensive monitoring because of ventricular proarrhythmia risk, which restricts their use to specialized settings and may exacerbate disparities in access to care. Development of atrial-selective antiarrhythmic therapies could enable safer outpatient rhythm-control strategies and expand access to rhythm management across diverse healthcare environments. In this context, the atrial-specific potassium channel Kv1.5 represents a promising therapeutic target to bridge the gap between clinical necessity and equitable care.
2. Kv1.5 Channels and Atrial Electrophysiology
Voltage-gated potassium channels are key determinants of cardiac action potential duration by regulating membrane repolarization through multiple potassium currents [32,33]. Among these, the Kv1.5 channel, encoded by the KCNA5 gene, has received considerable attention due to its contribution to the ultra-rapid delayed rectifier potassium current (IKur) in the human heart [34]. IKur is the major repolarizing current in the atria; at the same time, it contributes minimally to ventricular electrophysiology [34,35]. Therefore, this atrial selectivity identifies Kv1.5 as a therapeutic target for the treatment of atrial fibrillation (AF) [36].
2.1. Contribution of Accessory Subunits to Kv1.5 Channel Behavior
Kv1.5 channels form tetrameric complexes made of four pore-forming α-subunits, where auxiliary β-subunits modify how these channels open, close, and appear on the cell surface. Like many Kv channels, Kv1.5 is formed by four identical α-subunits, which form a functional potassium-selective pore [37]. There are several types of Kv β-subunits (Kvβ1–Kvβ3) that bind to Kv1 α-subunit subfamilies, and they have been shown to significantly modify the properties of Kv channels. Kv β-subunits can modulate Kv1.5 channel assembly, intracellular trafficking, and gating kinetics, as well as serve as a metabolic sensor of the cell (i.e., NAD(P)H) [33,38]. For example, Kv β-subunits act as chaperones to help proper folding and surface expression at the plasma membrane of Kv1.5 channels, and the N-terminus of Kv β-subunits has been demonstrated to rapidly inactivate Kv1.5 currents (analogous to “N-type” inactivation). Furthermore, Kv β-subunits have an enzymatic cleft that binds NADPH/NADP+, and changes in the redox state will affect how Kv β-modulates Kv1.5 currents, thus providing a mechanism for the channels’ behavior to be linked to cellular metabolic signals [38]. Therefore, through these mechanisms, Kv1.5 auxiliary β-subunits refine the function of Kv1.5 channels in electrically active tissues such as the heart [33,38].
2.2. Kv1.5 Channels as Promising Targets for Antiarrhythmic Drug Development
Early interest in Kv1.5 as an antiarrhythmic target grew from an atrial-predominant expression pattern. Studies show high Kv1.5 and IKur expression in human atrial myocardium, with minimal or functionally limited presence in ventricular tissue [34,35]. This demonstrates a clear contrast with other repolarizing potassium currents, including IKr and IKs, that support ventricular repolarization and are often linked to drug-induced ventricular arrhythmias [39]. In fact, blockade of IKr (hERG channels) by class III antiarrhythmic agents or other drugs often prolongs the QT interval and can precipitate torsades de pointes and ventricular fibrillation in susceptible patients [39]. This atrial-selective distribution led researchers to view IKur inhibition as a strategy to alter atrial electrophysiology while leaving ventricular repolarization largely unchanged, therefore avoiding ventricular proarrhythmia [33].
Functionally, IKur drives early repolarization and helps set the action potential duration and effective refractory period [33,37]. By shaping the plateau and repolarization phases of the atrial action potential, IKur affects the atrial tissue’s capacity to support rapid or re-entrant electrical activity [40]. Pharmacological inhibition of Kv1.5 prolongs atrial action potential duration and increases effective refractory period, which lowers the likelihood that atrial myocardium sustains the re-entrant circuits underlying atrial fibrillation [41,42]. Because IKur shows minimal involvement in ventricular repolarization, this prolongation stays largely restricted to atrial tissue, at least in theory [41].
Atrial selectivity has major clinical value during antiarrhythmic drug development. Many antiarrhythmic drugs for atrial fibrillation fail due to safety concerns rather than weak efficacy, often driven by ventricular proarrhythmia, QT interval prolongation, and extracardiac toxicity [36]. Class III antiarrhythmic agents illustrate this problem clearly; they act on ventricular potassium currents and raise the risk of torsades de pointes and sudden cardiac death in susceptible patients [39]. This risk profile limits long-term use in clinical practice. Against this background, researchers focused on atrial selective targets such as Kv1.5, which was seen as a potential solution aimed to separate rhythm control benefits from ventricular risk and improve safety for patients with atrial fibrillation [41].
These factors drove extensive preclinical and early translational work focused on Kv1.5 and IKur inhibitors as atrial selective antiarrhythmic agents [43]. Early electrophysiology studies showed that selective Kv1.5 blockade prolongs atrial refractoriness without triggering ventricular arrhythmias, providing optimism for safer rhythm-control strategies in patients with atrial fibrillation [42]. Despite a strong biological rationale, translation of Kv1.5 targeted therapies into routine clinical care has remained limited. To date, the only Kv1.5 IKur targeting drug to reach clinical use is vernakalant, which was approved in Europe for acute cardioversion of AF and has mixed ion channel effects (including atrial-selective Na+ and K+ current blockade) [44]. This gap in therapies arises from challenges beyond target identification alone. Issues such as drug pharmacokinetics, atrial remodeling in persistent AF, and off-target effects have also posed hurdles. The sections ahead review the molecular control of Kv1.5, results from preclinical and early clinical studies, cardiac trials of Class III Antiarrhythmics, and safety side effects and drug-drug interactions of Kv1.5 inhibitors and class III antiarrhythmics.
3. Molecular Regulation of Kv1.5 Channels in Health and Disease
Atrial-predominant Kv1.5 expression supported the idea of atrial selective antiarrhythmic therapy. Unlike in ventricles, robust Kv1.5 currents in atria allow pharmacological blockade to affect atrial refractoriness with minimal ventricular proarrhythmia [45]. However, the Kv1.5 function does not stay fixed over time. Kv1.5 activity shifts through regulation at several molecular levels, including subunit composition, channel trafficking, and sensitivity to the cellular redox environment. These mechanisms regulate the number of channels that reach the cell membrane and their activity during electrical signaling [46]. This is important in atrial fibrillation, where atrial myocardium undergoes marked electrical, structural, and metabolic remodeling [47,48]. During this disease state, changes in Kv1.5 regulation reshape atrial electrophysiology and may alter how atrial tissue responds to targeted therapies.
3.1. Kvβ1.2 Association with Kv1.5 Channel Complexes
Four α-subunits of KCNA5 assemble into Kv1.5 channels, yet how they function and reach the cell surface depends heavily on helper β-subunits [49]. One key player among them is Kvβ1.2, which can associate with Kv1.5 α-subunits and shapes Kv1.5 channel behavior. When Kvβ1.2 binds to the main units, it shifts with channel opening and closing, helps stabilize structures, and guides where the channel settles in the membrane [49].
3.2. Redox Regulation and Metabolic Sensing
What makes Kvβ1.2 stand out is that Kvβ1.2 (like other Kvβ subunits) contains an aldo-keto reductase domain that binds the redox cofactors NAD(P)H [50]. Through this mechanism, Kvβ1.2 provides Kv1.5 with metabolic sensing capability. The binding of pyridine nucleotides to Kvβ1.2 induces conformational changes that modulate Kv1.5 gating, effectively turning Kv1.5 into a sensor that connects a cell’s chemical environments to its electrical signals [51]. Elevated ratios of reduced cofactors (high NADH/NAD+ or NADPH/NADP+) favor more rapid Kv1.5 inactivation and decreased current, whereas oxidized conditions stabilize the open state [51]. The shifts in energy levels inside the cell affect the number of available channels and their output [51], thus tying electrical behavior to metabolism.
Regulatory β-subunits have also been shown to influence remodeling-relevant endpoints such as cell proliferation (Figure 1). In prior work from our group, stable co-expression of Kv1.5 with the Kvβ1.2 subunit (but not Kvβ1.1) was associated with a marked reduction in proliferation in a heterologous expression system (HEK293T cells). Image-based confluency analysis and cell counting reported markedly lower proliferation in Kv1.5+ Kvβ1.2 cells across 24–96 h [52].
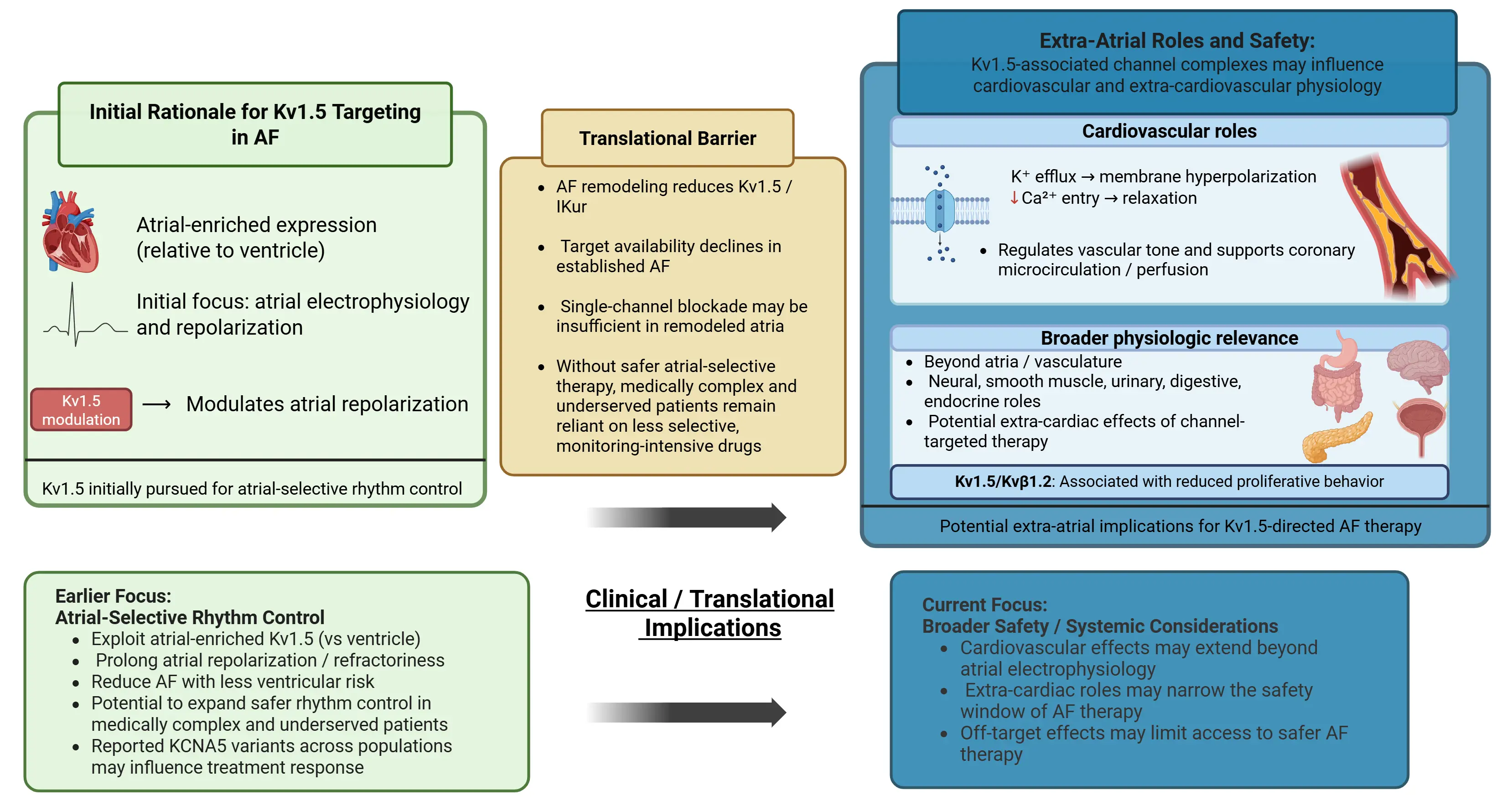
Figure 1. Kv1.5 in atrial fibrillation: initial rationale, translational barriers, and broader extra-atrial safety implications. Abbreviations: AF, atrial fibrillation; IKur, ultrarapid delayed rectifier potassium current. Created in BioRender. Molitor, F. (2026). https://BioRender.com/ktijtu2, accessed on 26 April 2026.
3.3. Oxidative Stress and Reactive Oxygen Species (ROS)
When cells face extra strain, such as during heart rhythm disorders (e.g., tachyarrhythmia or AF), small internal changes begin to occur. Oxidative stress shifts how Kv1.5 channels behave. Excess production of reactive oxygen species (ROS) can modify Kv1.5 channels and alter their function even in the absence of changes in gene expression [53]. Under oxidative conditions, Kv1.5 is subject to post-translational oxidative modifications (such as S-sulfenylation of cysteine residues) that reduce channel current and surface expression [53]. Experimental studies show that exposure to oxidants, such as hydrogen peroxide, alters both Kv1.5 expression and current strength in atrial muscle cells [54]. When reactive oxygen species are reduced through enzyme inhibition or antioxidant treatment, these effects lessen [55]. Compared with many other cardiac potassium channels, Kv1.5 responds more strongly to redox imbalance, producing a disproportionately strong reduction of Kv1.5-mediated current [47]. This marked sensitivity links cellular oxidative stress directly to atrial electrical behavior, as elevated ROS in AF can shorten atrial action potentials and promote arrhythmogenic substrate via Kv1.5 downregulation [53,56].
3.4. Angiotensin II (Ang II) and Nox4 Signaling Pathways
NADPH oxidase, specifically the form known as Nox4, is an upstream regulator of ROS-related Kv1.5 modulation. In atrial cardiomyocytes, exposure to angiotensin II turns on Nox4, which raises levels of ROS production that feed into Kv1.5 regulation [57]. Ang II signaling through AT1 receptor activates pathways including TGF-β1/Smad2/3 and ERK1/2, which increase Kv1.5 channel expression and stability, usually by upregulation of scaffolding proteins like SAP97 [58]. This Ang II effect is ROS-dependent; in cultured atrial myocytes, angiotensin II causes a rise in Nox-4-generated ROS, which in turn is required for the Smad2/3 and ERK-mediated increase in Kv1.5 levels [57]. When Nox4 activity is inhibited, it prevents the Ang II-induced upregulation of Kv1.5 [57]. For example, silencing Nox4 or supplementing cells with a reducing agent abolishes the Ang II-triggered increase in ROS and blocks the associated elevation in Kv1.5 expression and current [57]. Consistent with this, in vivo models there is a heightened angiotensin activity that drives parallel increases in Nox4, ROS, and Kv1.5 in the atria, whereas things such as AT1 receptor blockers or ROS scavengers can suppress these [57]. This Nox4-ROS-Kv1.5 interconnected signaling is especially pertinent to AF, which is often characterized by an overactive renin angiotensin system and oxidative injury in the atria [58,59]. Such results clearly point to an interconnected process linking hormonal factors, cellular oxidation, and ion channel behavior [57,58]. This connection gains importance in atrial fibrillation, a condition often marked by heightened renin-angiotensin system function and excess oxidative damage [60].
3.5. Nitric Oxide and eNOS Dysfunction
Control of Kv1.5 through redox process connects tightly to nitric oxide pathways, along with actions of endothelial nitric oxide synthase (eNOS). Under physiological conditions, eNOS produces nitric oxide, which supports vascular stability and limits inflammatory signaling. AF and oxidative stress can disrupt this balance. When ROS levels are high, eNOS becomes “uncoupled” due to oxidation or depletion of its essential cofactor (tetrahydrobiopterin, BH4) and substrate (L-arginine) [61]. An uncoupled eNOS no longer efficiently generates NO; instead, it preferentially diverts electrons to molecular oxygen to produce superoxide anion (O2•−) [61]. Such a switch intensifies oxidative damage, promoting issues in blood lining cells plus structural changes in heart chambers. In AF, for example, diminished NO bioavailability and increased superoxide in the left atrium have been documented [62]. Restoring redox balance alters the atrial cellular environment in which ion channels operate. These changes may indirectly influence the electrophysiological substrate associated with atrial fibrillation [63]. These effects show a link between redox homeostasis and tissue stability.
3.6. Mitochondrial Kv1.5 and Cellular Survival
Aside from appearing at the outer cell boundary, Kv1.5 channels also occur inside specific cells, such as within energy-producing structures known as mitochondria. Within these organelles, the presence of Kv1.5 appears to be tied to control mechanisms that regulate electrical balance across their membranes, reactive oxygen species production, and signals involved in programmed cell death. In pulmonary arterial smooth muscle, for example, a mitochondrial ROS–HIF-1α–Kv1.5 axis serves as an O2-sensing mechanism that couples changes in mitochondrial redox status to Kv1.5 activity and vascular tone [64]. Generally, mitochondrial redox status regulates Kv1.5 activity and cell fate decisions. Reduced mitochondrial ROS suppresses Kv1.5 expression and activity, contributing to mitochondrial and apoptosis-resistant phenotype, whereas preservation of Kv1.5 activity is associated with pro-apoptotic signaling in pulmonary arterial smooth muscle cells [64]. Evidence indicates this internal form might be associated with molecules that promote cellular breakdown, altering cell survival. In cancer cells, Kv1.5 expression is transcriptionally repressed (via HIF-1α and NFAT signaling), which helps the cells evade apoptosis [64]. Restoration of Kv1.5 in these cells, for instance, by using dichloroacetate (DCA) to inhibit mitochondrial pyruvate dehydrogenase kinase, reestablishes a mitochondria-to-Kv1.5 apoptotic feedback loop, inducing cancer cell death without harming normal cells. These findings position Kv1.5 within a feedback loop linking mitochondrial function, oxidative stress, and ion channel regulation. While the relevance of mitochondrial Kv1.5s’ exact importance in atrial cardiomyocytes remains unknown, these findings show important considerations regarding unintended consequences when blocking Kv1.5 in non-cardiac tissues. Notably, therapies targeting Kv1.5 (such as IKur blockers for AF) might have off-target effects in non-cardiac tissues where Kv1.5 modulates cell survival and metabolism.
It becomes clear that managing Kv1.5 in atrial fibrillation involves intricate layers. While initial hopes rested on its presence mainly in atria, activity of this channel shifts due to subunit interactions, changes driven by oxidation, and mitochondrial signaling, showing the complexity of managing Kv1.5 in chronic AF. All these elements can strongly influence Kv1.5 targeted therapies and provide context for the preclinical and clinical trials discussed in subsequent sections.
4. Preclinical Evidence for Kv1.5-Targeted Therapies
The biological case for atrial selective Kv1.5 inhibition drove extensive preclinical testing of IKur blockers as rhythm control therapies for atrial fibrillation. Experimental work addressed these questions across isolated atrial cells, perfused tissue preparation, and large animal models. Results across these systems supported the core concept behind atrial selective targeting. The same studies also exposed limits related to efficacy, safety margins, and translational consistency. These early findings provided compelling proof of concept evidence, while also revealing limitations that would later complicate clinical translation.
4.1. Early In Vitro and Electrophysiological Evidence
Research conducted early in vitro, using heterologous expression systems and isolated atrial cardiomyocytes, demonstrated that pharmacological blockade of Kv1.5 effectively decreases IKur and increases the duration of atrial action potentials [65]. Moreover, these effects were seen under normal physiological conditions, suggesting it would be possible to target IKur to increase atrial refractoriness, without creating ventricular electrical instability [66,67].
4.2. Animal Models and Proof of Atrial Selectivity
The experimental Kv1.5 inhibitor that has been the subject of the most extensive studies in preclinical atrial fibrillation models is DPO-1. Studies in primate and canine models showed that administration of DPO-1 to animals in vivo selectively lengthened the atrial effective refractoriness period with no significant effects on ventricular refractoriness or QT interval [68,69]. In an African green monkey model, the increase in atrial refractoriness with DPO-1 correlated with higher Kv1.5 expression in atrial vs. ventricular myocardium. These results support the concept of atrial selective modulation of electrophysiology by selective inhibition of IKur. Therefore, the selective inhibition of IKur could potentially be used as an anti-atrial fibrillatory agent with minimal proarrhythmic risk to the ventricles [68].
4.3. Additional Kv1.5 Inhibitors and Consistent Findings
Consistent with these findings, additional structurally distinct Kv1.5 inhibitors supported the atrial selective efficacy of IKur blockade. Two compounds, a triarylethanolamine (TAEA) and an isoquinolinone derivative (ISQ-1), were shown to potently inhibit human Kv1.5 currents expressed in heterologous systems and to selectively prolong atrial refractoriness in vivo without affecting ventricular refractoriness or QTc interval. In anesthetized and conscious canine models, both compounds had increased atrial ERP, prolonged atrial conduction intervals, and terminated inducible atrial fibrillation, while also sparing ventricular electrophysiology [69].
4.4. Limitations: Remodeling and Substrate Dependence
Preclinical studies also validated the atrial selective safety profile of kur inhibition. Individual Kv1.5 blockers, DPO-1, XEN-D0101, and AVE0119, were all constantly found to cause dose-dependent, constant prolongation of action potential duration and ERP in models of atrial fibrillation, with no impact on ventricular repolarization or any proarrhythmic impact on the ventricle [70]. AVE0118, for example, was shown to restore atrial refractoriness in dogs and goats of atrial fibrillation without any pro-arrhythmic impact on the ventricle [70], while DPO-1 and XEN-D0101 were found to prolong atrial ERP and suppress AF in dog models without resulting in any measurable QT prolongation [70]. These consistent findings reinforce the concept that selective IKur blockade could achieve meaningful atrial electrophysiologic modulation while preserving ventricular electrical stability under controlled experimental conditions [70].
Preclinical research has also included studies of other Kv1.5 inhibitors, such as MK-0448 and related drugs, in models of atrial fibrillation. While MK-0448 successfully terminated atrial fibrillation in dogs and prolonged atrial refractory periods under specific conditions [71], further studies indicated that the effect of inhibiting IKur is influenced by the type of atrial substrate being studied and the disease state. When atrial fibrillation is long-standing (sustained) or has been modified by electrical remodeling, the effectiveness of MK-0448 in prolonging action potentials was reduced, suggesting that drug efficacy may be limited when the therapy targets a mechanism of action that is significantly altered by the disease state [72]. In human atrial tissue studies, for instance, selective IKur block elevated the action potential plateau but did not prolong, or even shorten, the AP duration in non-remodeled atria, whereas in chronic AF samples, some AP prolongation was seen. These findings indicate that the effectiveness of Kv1.5 inhibition is reduced once atrial fibrillation has induced molecular and electrical remodeling [72]. In other words, an AF substrate altered by long-term electrical remodeling, oxidative stress, or impaired Kv1.5 channel trafficking is less susceptible to IKur blockade. This substrate-dependent response helps to explain why drugs targeting Kv1.5 show diminished efficacy as atrial fibrillation becomes more chronic or structurally remodeled.
Subsequent cellular and tissue-level studies also showed that Kv1.5 expression and function are modified through a variety of mechanisms during AF. Chronic AF is associated with downregulation of Kv1.5 expression (reducing IKur density), as well as changes in subunit composition, oxidative damage, and reduced channel trafficking to the membrane [53,73]. This reduces the amount of Kv1.5 available at the cell surface for repolarizing the atria or alters the role of Kv1.5 in repolarizing the atria [74]. As such, the potential therapeutic benefit of selectively blocking IKur may be lost as atrial fibrillation progresses, which may explain why IKur blockers have shown diminished effectiveness with the progression of atrial fibrillation [40,66]. These studies illustrated how Kv1.5 targeted therapy is dependent upon both the disease state (disease duration) and the atrial substrate; therefore, these factors limit its clinical applicability in treatment for patients with atrial fibrillation.
Despite these difficulties, early-phase human studies have offered some limited but useful insight into the safety of inhibiting Kv1.5. The atrial selective Kv1.5 inhibitor XEN-D0101 was developed for a Phase 1 clinical trial, which found that XEN-D0101 had a very good safety profile, with indications of having atrial electrophysiological effects (prolonged atrial action potential indices) without overt ventricular pro-arrhythmic effects [75]. In isolated human atrial tissues from sinus rhythm and AF patients, XEN-D0101 significantly elevated the atrial AP plateau and increased contractility, while having no effect on ventricular action potentials [75]. Consistently, in healthy volunteers, XEN-D0202 did not significantly prolong the QTc interval at doses up to 300 mg [75]. These findings confirm the atrial selective profile and acute safety of Kv1.5 blockade in humans [75]. Nonetheless, the lack of larger-scale efficacy trials and longer-term outcome data led to little further clinical advancement. Similar to other preclinical compounds previously discussed, uncertainty regarding its efficacy in remodeled atria and the limited clinical efficacy observed in subsequent testing were major factors leading to the compound never progressing to late-phase clinical trials [75].
4.5. Early Human Translational Evidence
Overall, the findings from both preclinical and early translational research clearly support the notion that Kv1.5 inhibitors can selectively modulate atrial electrical properties and inhibit atrial arrhythmias under controlled experimental conditions. Simultaneously, however, there were clear limitations as well, including substrate-dependent efficacy, limited effectiveness against atrial remodeling, and a lack of knowledge about long-term safety. Together, these limitations provide some insight into why Kv1.5-targeted treatments for arrhythmias have not yet been widely adopted in the clinic.
5. Cardiac Trials of Class III Antiarrhythmics (Kv Channel Blockers)
Pharmacologic rhythm control in AF relies on two main drug classes: Class Ic and Class III antiarrhythmic agents. Class Ic agents, including flecainide and propafenone, are widely used for maintaining sinus rhythm in patients without structural heart disease, reduced left ventricular function, or significant coronary artery disease, and are often preferred in younger, healthier patients due to their favorable tolerability profile [76]. Their use has traditionally been contraindicated in structural heart disease based on the CAST trial, which demonstrated increased mortality with Class Ic agents in post-myocardial infarction patients. However, emerging evidence suggests this risk may have been overgeneralized and that flecainide may be safe in carefully selected patients with stable structural heart disease [76]. Nevertheless, a substantial proportion of AF patients, particularly those with heart failure, CKD, or significant comorbidities, remain ineligible for Class Ic therapy, reinforcing the need for safer atrial-selective options such as Kv1.5 inhibition. In patients ineligible for Class Ic therapy, Class III antiarrhythmic agents remain the cornerstone of pharmacologic rhythm control despite well-recognized safety limitations.
Class III antiarrhythmic drugs prolong cardiac repolarization through potassium channel blockade, increasing atrial refractoriness, but their effects are not confined to atrial tissue, exposing patients to risks such as QT prolongation and ventricular proarrhythmia. Nevertheless, their clinical durability across diverse atrial substrates, including structurally and electrically remodeled atria, has sustained their widespread use in routine practice. Understanding the strengths and liabilities of Class III agents provides a necessary framework for evaluating why atrial-selective targets such as Kv1.5 have struggled to replace them.
5.1. Amiodarone
Amiodarone is a broad-spectrum antiarrhythmic agent that works through Class III effects as well as other mechanisms of action. It blocks multiple Kv channels in addition to Na+, Ca2+ channels, and β-adrenergic receptors [77]. Amiodarone is effective at maintaining sinus rhythm in atrial fibrillation and at reducing the occurrence of ventricular arrhythmias; however, the Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT) demonstrated no significant reduction in all-cause mortality with long-term amiodarone therapy compared with placebo in high-risk populations [78]. Overall, amiodarone is highly effective for refractory atrial and ventricular arrhythmias but is typically reserved for short-term or last-line use due to its toxicity burden [77].
5.2. Dronedarone
Dronedarone is an amiodarone analog, which is less lipid soluble than amiodarone and does not contain iodine [79]. These changes reduce dronedarone’s organ toxicities (thyroid and pulmonary) and shorten its half-life [79]. Like amiodarone, it’s a class III potassium channel blocker and has additional antiarrhythmic activity. In the ATHENA trial, dronedarone reduced the composite endpoint of cardiovascular hospitalization or death by approximately 24%, largely driven by fewer AF-related hospitalizations, leading to regulatory approval [80]. Additionally, dronedarone resulted in lower rates of arrhythmia-related death and stroke in AF patients over 21 months of follow-up (2.7% vs. 3.9%). This reflects a reduction in arrhythmic mortality, and there was no significant difference in overall mortality between groups [80]. However, subsequent trials identified important safety concerns: ANDROMEDA was terminated early due to increased mortality in patients with systolic heart failure [81], and PALLAS was halted after showing higher rates of stroke, heart failure, cardiovascular death, and all-cause mortality in patients with permanent AF [82]. Adverse outcomes in PALLAS were attributed in part to treatment of a higher-risk population and drug-drug interactions, particularly increased digoxin exposure. Although dronedarone appears to be beneficial in reducing these endpoints in AF patients, the benefit is specific to certain contexts. Compared with amiodarone, dronedarone is less effective at maintaining sinus rhythm. In the DIONYSOS trial, AF recurrence was higher with dronedarone, but it had fewer non-cardiac toxicities, particularly thyroid and neurologic effects [83]. Overall, dronedarone offers moderate rhythm-control efficacy with less organ toxicity than amiodarone, but its use is limited by HF risk and significant drug interactions [84].
5.3. Sotalol
Sotalol is a class III antiarrhythmic that blocks the rapid delayed rectifier potassium current with additional non-selective β-adrenergic blocking activity. It is used to maintain sinus rhythm in AF and to suppress certain ventricular arrhythmias. Compared with amiodarone and dronedarone, it is a more selective Kv blocker, with β-blockade providing rate control and partial protection against excessive QT prolongation [85]. However, in a trial of pure K+ blocking sotalol (the dextro isomer without beta blocking), safety concerns were revealed. Most of the excess deaths were caused by sudden cardiac death due to arrhythmia [86]. These findings show that a pure class III drug can be pro-arrhythmic in patients at risk, SWORD was halted early because it resulted in harm [86]. Today, conventional racemic sotalol is still used with caution in patients. The beta blocking component in d,l-sotalol may decrease some risk; it does require monitoring of QT interval prolongation.
5.4. Dofetilide
Dofetilide is a class III antiarrhythmic, selective IKr/Kv11.1 blocker approved for conversion to and maintaining sinus rhythm in AF or atrial flutter. Its efficacy and safety in patients with structural heart disease, particularly those with left ventricular dysfunction, were demonstrated in the DIAMOND trial, which showed significantly higher rates of conversion to and maintenance of sinus rhythm compared to placebo, reduced heart failure hospitalizations, and no increase in overall mortality [87]. In the Danish DIAMOND trials (in patients with severe left ventricular dysfunction), dofetilide had no effect on all-cause mortality compared to placebo, but was antiarrhythmically effective [88]. Although mortality was not affected, those who did restore their sinus rhythm were found to have a significantly better survival (risk of death reduced by 56% in that subgroup) [88]. Dofetilide also has an effect on reducing hospital re-admissions: in DIAMOND, patients treated with dofetilide had a 30% reduction in all case readmissions (RR 0.70, p = 0.005), and a 31% reduction in heart failure-related readmissions compared to placebo [88]. These results show that dofetilide can be used relatively safely for AF even in high-risk patients if the QT interval is properly monitored, improving rhythm control and reducing morbidity, even if it does not improve longevity [88].
5.5. Ibutilide
Ibutilide is an intravenous class III antiarrhythmic for cardioversion of atrial fibrillation or flutter. Patients are given IV while in the hospital to convert sinus rhythm without electrical cardioversion. While this is effective for most patients, ibutilide carries a risk of polymorphic ventricular tachycardia (torsades de pointes) due to QT prolongation, making it so patients must be monitored continuously during administration [89]. Protocols advise monitoring for at least 4 h post infusion (or until QT return to baseline), and readiness with IV magnesium, defibrillator, and pacing in case torsades occurs [89].
5.6. Bretylium
Bretylium, an older class III K+ channel blocker (and sympatholytic) that was utilized in refractory ventricular fibrillation, especially in the pre-hospital or emergency settings prior to the use of amiodarone. A small, randomized trial in 1981 demonstrated that the addition of bretylium during CPR improved resuscitation rates [90]. These early results led to the identification of bretylium as a drug that could improve survival in victims of cardiac arrest [90]. Bretylium was referenced in early Advanced Cardiac Life Support (ACLS) protocols in the 1980s for refractory ventricular fibrillation, but its use gradually decreased when additional randomized studies highlighted the benefits of amiodarone for post-shock ventricular fibrillation. Bretylium is no longer readily available in most regions today, although it is an important historical example of a potassium channel blocker that was used in the acute care setting.
5.7. Vernakalant
Vernakalant is a relatively atrial-selective antiarrhythmic developed for rapid pharmacologic cardioversion of atrial fibrillation. Its atrial selectivity is driven primarily by blockade of potassium currents predominantly expressed in atrial tissue, with additional frequency-dependent sodium-channel effects and minimal impact on ventricular repolarization at normal heart rates. Intravenous vernakalant is approved in Europe and other regions for acute conversion of recent-onset AF (≤7 days) in non-surgery patients and less than 3 days in postoperative AF [91]. Conversion is rapid, typically occurring within 10–15 min in responders. Vernakalant’s atrial-selective profile is associated with a low risk of ventricular pro-arrhythmia, and torsades de pointes was not observed in clinical trials [91]. Vernakalant has been used clinically in Europe and Canada for over a decade but remains unapproved in the US following safety concerns raised during U.S. phase III trials [91,92]. Vernakalant occupies a niche role as a fast-acting, atrial-selective agent for acute AF cardioversion, but is geographically limited in availability.
Collectively, as outlined in Table 1, currently available Class III agents trade atrial efficacy for systemic toxicity, proarrhythmia risk, or intensive monitoring requirements.
Table 1. Past and Present Medication Use of Class III Antiarrhythmic Drugs.
|
Use |
Use/Indication |
Key Trial Finding |
|---|---|---|
|
Bretylium (Class III + sympatholytic) |
Refractory VF/VT (historic) |
IV bretylium improved survival to hospital admission during cardiac arrest (35% vs. 6% placebo; p < 0.05) but has been replaced by amiodarone in modern practice [90,93]. |
|
Amiodarone (Class III, plus Class I/II/IV) |
AF maintenance; VT/VF suppression |
SCD-HeFT: no survival benefit vs. placebo (all-cause death 28% vs. 29%; HR ≈ 1.06) despite high arrhythmia suppression; long-term use limited by toxicity. Whereas implantable cardiovascular defibrillator (ICD) decreased mortality by 23% [78,94]. |
|
Sotalol (d,l-isomer; Class III + β-blocker) |
AF maintenance; limited VT suppression |
SWORD (d-sotalol): increased mortality post-MI with LV dysfunction (5.0% vs. 3.1%; RR 1.65). Racemic sotalol remains in use with telemetry-guided initiation [86]. |
|
Dofetilide (Class III, IKr) |
AF/AFI conversion & maintenance; safe in HF/CAD |
DIAMOND-AF: sinus rhythm restoration 59% vs. 34% placebo; 1-yr maintenance 79% vs. 42% (p < 0.001). No mortality difference: inpatient QT monitoring required [88]. |
|
Ibutilide (Class III; late INa effect) |
IV cardioversion of recent-onset AF/AFI |
Converts ~30–50% of AF and ~50–70% of atrial flutter; significant QT prolongation and torsades risk necessitate continuous ECG monitoring [89,95]. |
|
Vernakalant (Atrial-selective, multichannel) |
Rapid IV cardioversion of recent-onset AF |
ACT I–IV/AVRO: conversion ~44.9–51.7% within 90 min (often within 10–15 min in responders); superior to IV amiodarone at 90 min (51.7% vs. 5.2%, p < 0.0001); superior to IV amiodarone; low ventricular proarrhythmia risk; approved in Europe, not US [96,97,98]. |
|
Dronedarone (Class III, multichannel) |
AF/AFI (non-permanent); rhythm control without severe HF |
ATHENA: Dronedarone reduced the composite endpoint of cardiovascular hospitalization or death versus placebo (31.9% vs. 39.4%; HR 0.76, p < 0.001), driven primarily by fewer AF-related hospitalizations, supporting regulatory approval [80]. |
Abbreviations: AF = atrial fibrillation; AFI = atrial flutter; VF = ventricular fibrillation; VT = ventricular tachycardia; MI = myocardial infarction; LV = left ventricular; HF = heart failure; CAD = coronary artery disease; SCD = sudden cardiac death; ICD = implantable cardioverter-defibrillator; ECG = electrocardiogram; IV = intravenous; HR = hazard ratio; RR = relative risk; IKr = rapid delayed rectifier potassium current; late INa = late sodium current.
6. Safety, Adverse Effects, and Drug–Drug Interactions
6.1. Safety of Kv1.5 Inhibitors
Kv1.5 channel blockers (IKur inhibitors) represent a newer class of investigational antiarrhythmic drugs designed to selectively prolong atrial repolarization while not affecting ventricular electrophysiology. Consistent with this, early human electrophysiology studies of the IKur inhibitor XEN-D0103 showed that, even at a high plasma concentration (10 µM), it had no significant effects on ventricular action potentials and caused no significant QTc prolongation in healthy volunteers [99].
Despite this, the potential for small-molecule Kv1.5 inhibitors to produce non-selective effects on cardiac K+ channels remains a significant concern regarding safety. For example, many of these small molecules will inhibit additional cardiac K+ currents at higher concentrations than their intended target. For instance, the prototype IKur blocker DPO-1 has an IC50 of only 30 mM for Kv1.5; however, it can begin to affect the transient outward current (Ito) and potentially the rapid delayed rectifier (IKr) in the micromolar concentration range [70]. Likewise, XEN-D0103 inhibits Kv1.5 with an IC50 ≈ 25 nM and exhibits > 500-fold selectivity against hERF (IKr) and several other cardiac channels [70]. The unintentional blockade of hERG with drugs, such as those that inhibit Kv1.5, could potentially eliminate atrial selectivity and contribute to prolonged ventricular repolarization if excessive drug levels occur. In fact, one of the primary design objectives was to establish a significant safety margin between the drug concentrations required to inhibit IKur and drug concentrations required to inhibit hERG [70]. Fortunately, XEN-D0101 demonstrated an IC50 of 13 µM against hERG (vs. 0.24 µM for Kv1.5) and did not demonstrate any QTc prolongation at doses of 300 mg or less in humans, indicating a large safety margin [75]. Ultimately, although careful medicinal chemistry has significantly enhanced the selectivity of IKur inhibitors, the liability of hERG inhibition and the risk of off-target K+ blockade still require monitoring during the development phase to prevent compounds from causing the very arrhythmias they were designed to treat [75].
So far, side effects associated with IKur inhibitors have been generally milder than would be expected based on the amount of time these drugs have been in use as medications in humans. During an initial trial of MK-0448 (an IV-administered Kv1.5 blocker), participants experienced only mild side effects from the treatment and had very few issues when they received the treatment by vein through the IV line, the most commonly noted issue being minor redness at the site where the IV was administered. There were no serious heart rhythm-related complications reported in this study, nor were there any organ toxicity problems noted either [71]. Similarly, XEN-D0101, administered orally, did not produce any statistically significant QTc prolongation or hemodynamic changes up to its highest tested dose (300 mg) [75]. These initial safety signals are promising, but experience is limited to small phase I trials in healthy subjects. Potential side effects outside the heart are still unknown, as Kv1.5 channels are present in other tissues such as immune cells, the kidneys, skeletal and vascular smooth muscle, and, to a lesser extent, the brain [100], so off-target blockade could theoretically produce effects like mild vasomotor changes or central nervous system effects, though none have emerged so far. In summary, recent data do not link current inhibitors to multi-organ toxicities as was observed with previous drugs (e.g., amiodarone), though a full assessment of toxicities will require larger and longer trials.
Drug-drug interactions with Kv1.5 inhibitors are not yet defined, as these agents are still investigational. They are expected to be strong inhibitors or inducers of CYP450 enzymes, and, so far in preliminary trials, there have been no reports of clinically relevant PK interactions. However, a cautious strategy is to be proactive in considering interactions. Because IKur blockers are intended to be atrially selective, it may be possible to combine them with other antiarrhythmics. For example, preclinical data in a goat model of chronic AF show that adding the Kv1.5 blocker AVE0118 to the class III drug (dofetilide or ibutilide) markedly improves AF termination without prolonging the QT interval [101]. In that study, AVE0118, along with prolonged atrial refractoriness, and when followed by dofetilide or ibutiide all animals were cardioverted to sinus rhythm (versus only 20% conversion with dofetilide/ibutilide alone) [101]. This indicates a positive synergistic effect in which an atrial selective drug produces incremental benefit without increased ventricular proarrhythmia. However, the co-administration of multiple channel blockers in real patients should still be done with caution. Until extensive clinical experience has been gained, it is prudent to avoid combining IKur inhibitors with other drugs known to prolong the QT interval, or to limit such combinations to settings where they have been closely monitored. Even a drug with excellent Kv1.5 selectivity may still have subtle hERG blocking effects, and this activity may be exacerbated by other IKr blockers, potentially unmasking arrhythmias. In summary, while no specific contraindicated combinations have yet been established for Kv1.5 blockers, cautious guidelines (avoiding overlaps with other antiarrhythmics or with CYP3A4 substrates unless monitored carefully) should be followed. As these drugs are advanced through the trial process, formal studies of drug-drug interactions will establish their metabolism and identification behavior in clinical practice.
6.2. Class III Antiarrhythmic Drug Effects and Proarrhythmia Risk
Class III antiarrhythmic drugs exert their antiarrhythmic effect by prolonging cardiac repolarization; they block the potassium currents responsible for phase 3 repolarization [102,103]. Consequently, QT prolongation is a class effect. This lengthens the ventricular repolarization (QT interval) on the ECG [102]. Minor QT prolongation can be therapeutically beneficial by preventing mature beats, but excessive QT prolongation can lead to early afterdepolarizations and torsades de pointes [104]. All Class III agents are pro-arrhythmic but differ in torsadogenic risk. Pure IKr antagonists (dofetilide, IV ibutilide) have among the highest torsades risks, necessitating monitored initiation [104,105]. Dofetilides clinical trials reported torsades in roughly 0.9–3.3% of patients (mostly within the first three days of therapy) under monitored initiation [104]. Sotalol, which blocks Ikr and has beta-blocking activity, has an approximately 1% incidence of torsades at recommended doses (higher with supratherapeutic dosing or in renal impairment) [27]. Accordingly, both sotalol and dofetilide must be initiated in the hospital with ECG monitoring to mitigate this risk [27,104]. Ibutilide, administered intravenously for acute cardioversion of atrial fibrillation and atrial flutter, also carries a risk of torsades de pointes. In a pooled analysis of 1720 patients, torsades occurred in 3.9% of cases, typically during or within 45 min of infusion [105]. This study also showed that ibutilide induced torsade de pointes in 5.6% of 304 women and 3% of 792 men (p = 0.05), suggesting a greater incidence in women than in men [105]. In contrast, amiodarone, despite extreme QT prolongation, has a notably lower incidence of torsades (<1%) [106]. This is attributable to amiodarone’s “multifaceted” electrophysiological properties: it blocks inward Na+ and Ca2+ currents and has beta-adrenergic blocking activity [103], all of which suppress the EADs and dispersion of repolarization, mitigating the arrhythmogenic effect of QT prolongation [103]. Yet caution for amiodarone is still justified as it can induce torsades in “high risk” scenarios (severe hypokalemia or concomitant QT prolonging drugs), so electrolyte and drug interaction monitoring is required. Finally, bretylium (an older class III agent rarely used) also blocks K+ channels and prolongs ventricular action potential duration. There are sparse clinical data on bretylium’s risk for torsades due to this drug’s niche use, but this drug’s initial sympathomimetic activity (by triggering norepinephrine release) can elicit ventricular ectopy with its use [107]. Also, hypotension (rather than torsades) is the most common acute adverse effect of bretylium [107].
As shown in Table 2, these adverse-effect profiles differ qualitatively across agents, with some drugs limited primarily by ventricular proarrhythmia and others by cumulative systemic toxicity or hemodynamic intolerance.
Table 2. Major Adverse Effects of Class III Antiarrhythmics.
|
Drug |
Major Adverse Effects |
|---|---|
|
Bretylium |
Profound hypotension (often supine; postural hypotension common; ~50%); transient ↑BP/arrhythmias early via NE release; N/V (esp. rapid IV); flushing/headache [108]. |
|
Amiodarone |
Multi-organ toxicity: pulmonary pneumonitis/fibrosis (~5–7%) [109] thyroid (hypo/hyper); hepatic injury; skin/ocular (blue-gray skin, corneal deposits); neurologic (neuropathy, tremor). Cardiac: bradycardia/AV block; QT prolongation (TdP risk low <1%) [110]. |
|
Sotalol |
β-blockade: bradycardia, hypotension, fatigue; bronchospasm (reactive lung disease); exercise intolerance; AV block. Proarrhythmia: QT prolongation/TdP ~1–5% (↑ dose/renal failure) [27]. |
|
Dofetilide |
Dose-dependent QT prolongation/TdP (primary); minimal hemodynamic effects (little HR/BP change); occasional headache/dizziness/GI upset; requires initiation of ECG monitoring [104,111]. |
|
Ibutilide |
Marked QT prolongation; TdP risk up to 8.3% (↑ with underlying cardiomyopathy) [104,112]. requires continuous ECG monitoring (≥4 h) [89]. Transient hypotension/bradycardia during infusion; hypoK/hypoMg increases risk [95]. |
|
Vernakalant |
Acute infusion reactions: hypotension and bradycardia; rare ventricular proarrhythmia reported in trials (TdP not observed in major studies). Minor: dysgeusia, sneezing. Avoid/contraindicated in hemodynamic instability, severe HF, acute coronary syndromes, or hypotension [96]. |
|
Dronedarone |
Less organ toxicity vs amiodarone; GI upset (nausea/diarrhea); transient LFT elevations [113]; rare pulmonary/chest pain; monitor for HF decompensation (contraindicated in advanced HF); fewer thyroid/ocular effects than amiodarone [114]. |
Abbreviations: AV = atrioventricular; BP = blood pressure; ECG = electrocardiogram; GI = gastrointestinal; HF = heart failure; HR = heart rate; IV = intravenous; LFT = liver function tests; N/V = nausea/vomiting; NE = norepinephrine; TdP = torsades de pointes; ↑ = increased; hypoK = hypokalemia; hypoMg = hypomagnesemia.
Beyond repolarization, off-target cardiac effects of class III drugs significantly shape their safety profiles. Importantly, most are not purely selective IKr blockers. For example, Amiodarones class I (Na+) and class IV(Ca2+) effects, as well as beta-blockade [103], show why these drugs treat multiple arrhythmias and why you see side effects. Drug-specific nonarrhythmic adverse effects across Class III agents are summarized in Table 2. Ibutilide has another effect that is unrelated to other drugs. It enhances a slow inward Na+ current into the cell at the end of the action potential (phase 3), therefore prolonging repolarization [115]. Although this enhances the ability of ibutilide to prolong the action potential, it can also promote ventricular arrhythmias if used improperly [95]. Dofetilide is highly selective for IKr; thus, it does not directly affect Na+ or Ca2+ channels, resulting in little change in heart rate or blood pressure. The primary concern with dofetilide is related to its dose-dependent increase in QT interval and increased risk of torsades de pointes [104], which is why dosing is adjusted for renal function and monitored. Bretylium has a completely different set of off-target pharmacological properties. Initially, bretylium causes norepinephrine to be released from the nerve endings in the sympathetic nervous system. After this occurs, breylium causes a ganglionic blockade [107]. Thus, when breylium is first given, there is an initial hypertension, and occasionally there will be some arrhythmias immediately after administration of breylium. However, eventually, adrenergic tone drops, and therefore, there will be a significant drop in blood pressure (hypotension). Clinically, the most frequent immediate concern with the use of bretylium was hypotension, rather than torsades de pointes [116].
6.3. Drug–Drug Interactions
Drug-drug interactions are an important part of Class III drug safety, especially when they are used in polypharmacy situations with cardiovascular and noncardiovascular drugs. Specific interaction profiles and major contraindicated combinations are outlined in Table 3. These limitations highlight the narrow therapeutic window of conventional Class III antiarrhythmic therapy and explain why safety concerns, rather than lack of efficacy, often limit long-term use. These challenges have driven interest in atrial-selective strategies designed to preserve rhythm-control efficacy while minimizing ventricular proarrhythmia, systemic toxicity, and clinically significant drug–drug interactions.
Table 3. Drug-Drug Interactions for Class III Antiarrhythmics.
|
Drug |
Notable Interactions/Cautions |
|---|---|
|
Bretylium |
Catecholamine release may worsen digoxin toxicity; potentiated by MAO inhibitors; enhanced pressor responses to catecholamines → monitor BP; caution with other hypotensives (additive BP drop). (Obsolete; limited modern interaction data) [117]. |
|
Amiodarone |
↑ digoxin (~79%), rivaroxaban (~38%), phenytoin (~59%) [118]; ↑ warfarin effect/bleeding risk; additive QT prolongation (avoid macrolides/fluoroquinolones/antipsychotics/TCAs); with β-blockers or non-DHP CCBs ↑ bradycardia/AV block risk [119]. |
|
Sotalol |
Renal elimination; few PK interactions (no CYP). Avoid additive bradycardics and QT-prolongers (β-blockers, Ca2+ blockers, other Class III) → bradycardia/hypotension/TdP risk [27]. |
|
Dofetilide |
Renal cationic secretion: contraindicated with cimetidine, trimethoprim, verapamil, HCTZ, ketoconazole, prochlorperazine; avoid/contraindicated with renal cation secretion inhibitors (e.g., metformin, amiloride), CYP3A4 inhibitors (macrolides, azoles, amiodarone), and other QT-prolongers; caution with digoxin [120]. |
|
Ibutilide |
Hepatic metabolism (CYP450). Avoid coadministration with QT-prolongers (Class III agents, some antipsychotics/antifungals/macrolide-like agents) and electrolyte-altering drugs [95]. |
|
Vernakalant |
Limited interaction data; avoid combination with other QT-prolonging antiarrhythmics or drugs that increase TdP risk unless closely monitored. Use caution with AV-nodal–slowing agents (additive bradycardia/hypotension risk during infusion) [96]. |
|
Dronedarone |
Moderate CYP3A4 inhibitor; P-gp inhibitor → ↑ simvastatin, dabigatran, digoxin; contraindicated with strong CYP3A4 inhibitors/inducers; additive QT risk similar to other Class III agents [79]. |
Abbreviations: AV = atrioventricular; BP = blood pressure; CCB = calcium channel blocker; CYP = cytochrome P450; CYP3A4 = cytochrome P450 3A4; DHP = dihydropyridine; HCTZ = hydrochlorothiazide; MAO = monoamine oxidase; PK = pharmacokinetic; P-gp = P-glycoprotein; QT = electrocardiographic interval from the start of the QRS complex to the end of the T wave; TdP = torsades de pointes; TCA = tricyclic antidepressant; ↑ = increased.
7. Why Kv1.5 Has Not Translated to Effective Arrhythmia Therapy
Kv1.5 (IKur) has long been considered an attractive atrial-selective target because of its predominant expression in atrial myocardium and absence in ventricles, raising the possibility of suppressing AF without ventricular pro-arrhythmia. Several representative compounds illustrate this translational gap.
XEN-D0103 (Xention Ltd., Cambridge, UK) illustrates this challenge. Although the compound showed strong atrial selectivity in preclinical studies, a placebo-controlled clinical trial in patients with paroxysmal AF using continuous rhythm monitoring did not demonstrate a significant reduction in AF burden [70]. Similarly, MK-0448 (Merck, NJ, USA), another selective Kv1.5 inhibitor, advanced through early-phase testing and showed atrial electrophysiologic effects in preclinical models, but no convincing Phase II efficacy data in AF were reported, and development was not pursued further [121,122]. Broader atrial-selective or multichannel approaches have also fallen short: compounds such as AVE0118/AVE1231 did not progress beyond early development [70], and celivarone, a non-iodinated amiodarone analog with multichannel activity including atrial potassium currents, failed to improve sinus rhythm maintenance in Phase II trials and was discontinued [122]. To date, no Kv1.5-selective small-molecule antiarrhythmic has reached the market.
A central reason for these failures lies in a fundamental paradox in the biology of human AF: the very disease state that creates the need for Kv1.5-targeted therapy simultaneously erodes the molecular target itself. As described in Section 3, chronic AF drives substantial electrical remodeling of atrial tissue, including progressive downregulation of Kv1.5 expression and a corresponding reduction in IKur current density [30]. This process is accelerated by oxidative stress, Nox4-driven ROS production, and angiotensin II signaling, which characterize the remodeled atrium and alter Kv1.5 trafficking, subunit composition, and surface expression [47,57]. The result is a self-defeating therapeutic scenario: by the time a patient presents with established or persistent AF and requires rhythm control, the IKur current that selective blockers were designed to target has already been substantially reduced. Blocking a diminished current produces a correspondingly diminished electrophysiologic effect, which likely explains why drugs that showed compelling prolongation of atrial refractoriness in preclinical models and early-phase human studies failed to demonstrate meaningful AF burden reduction in larger trials [70,72]. Thus, chronic AF may render the Kv1.5/ IKur target functionally less available precisely when antiarrhythmic therapy is most needed.
The multifactorial nature of AF maintenance compounds this substrate-dependent limitation. Chronic AF is maintained through multiple overlapping mechanisms beyond IKur, including structural fibrosis, calcium handling abnormalities, gap junction remodeling, and autonomic dysregulation [123]. Targeting a single ion channel that is itself downregulated in the disease state, therefore faces a dual challenge: reduced target availability and insufficient impact on the broader arrhythmogenic substrate. Future strategies targeting Kv1.5 may need to account for disease stage, selecting patients earlier in the AF trajectory where IKur density remains sufficient for meaningful pharmacological modulation, or combining IKur blockade with therapies that address upstream remodeling mechanisms.
8. Arrhythmia from a Public Health Perspective
Cardiac arrhythmias, particularly AF, represent a growing public health challenge with implications that extend far beyond individual clinical encounters. Population-level data consistently show rising arrhythmia-related morbidity and mortality over recent decades, with atrial fibrillation and flutter-related deaths now surpassing those due to cardiac arrest in several regions, including the US. Projections suggest that, despite advances in cardiovascular care, the burden of AF-related mortality will continue to increase through 2040, especially among younger age groups and socioeconomically disadvantaged populations [124].
Globally, the prevalence, disability-adjusted life years (DALYs), and mortality associated with AF have risen steadily from 1990 to 2021, with further increases projected into the next decade [125]. Although higher-income regions currently report the greatest absolute AF burden, lower- and middle-sociodemographic index (SDI) regions are experiencing more rapid growth rates, likely reflecting population aging, rising cardiometabolic risk factors, and improved detection over time. Importantly, underdiagnosis remains a persistent concern in lower-SDI settings, where limited access to screening, ambulatory monitoring, and specialty care may obscure the true prevalence of AF and delay treatment until complications occur [125].
Disparities in arrhythmia outcomes are not limited to global differences. In the United States, racial and geographic inequities persist, with Black populations experiencing higher arrhythmia-related mortality despite lower rates of documented AF diagnosis, suggesting gaps in detection, access to care, or treatment quality [124]. Rural communities face additional challenges due to fewer electrophysiology centers, limited access to continuous ECG monitoring, and reduced availability of advanced therapies such as catheter ablation [72,126]. While ablation is now guideline-recommended for many patients with symptomatic AF, its dependence on specialized infrastructure restricts its use in low-resource settings, increasing reliance on pharmacologic rhythm or rate control.
Even among drug-based therapies, access and safety considerations shape public health impact. Certain antiarrhythmic agents, such as sotalol and dofetilide, require in-hospital initiation with continuous ECG monitoring to mitigate the risk of torsades de pointes [127], creating barriers for patients without reliable access to inpatient care. Policy changes can influence these barriers, in the US, the removal of the FDA Risk Evaluation and Mitigation Strategy (REMS) requirement for dofetilide in 2016 was intended to broaden access while maintaining safety through label-mandated monitoring [128,129].
The failure of atrial-selective potassium channel targets such as Kv1.5 to translate into effective therapies has left clinicians reliant on older drugs like amiodarone, with significant toxicity and monitoring burdens. These limitations disproportionately affect patients with multiple comorbidities and health systems with limited resources. Arrhythmia management, therefore, represents not just a clinical challenge, but a broader public health, a priority, one in which the development of atrial-selective agents such as Kv1.5 inhibitors could play a meaningful role by expanding safe rhythm-control options to patients currently excluded from existing therapies.
9. Kv Channel Modulation in Vascular Tone and Vascular Disease
While the primary focus of this review is atrial electrophysiology, the vascular expression of Kv1.5 is relevant to AF management because inhibition of Kv1.5 in coronary vessels could alter microvascular tone and myocardial perfusion, representing a potential off-target safety consideration for any Kv1.5-directed antiarrhythmic therapy. Beyond cardiac arrhythmias, Kv channels play an important role in regulating vascular smooth muscle tone and have been explored as potential therapeutic targets in vascular disease. Vascular smooth muscle cells express several Kv channel families, especially Kv7 (KCNQ), which help maintain the resting membrane potential [130]. The activation of these channels promotes membrane hyperpolarization, reduces calcium influx, and leads to vasodilation, whereas Kv inhibition favors vasoconstriction.
In the coronary microcirculation, Kv1.5 has been identified as a key mediator of metabolic dilation, the process by which coronary vessels dilate in response to increased myocardial oxygen demand [131]. Evidence from human microvessels indicates that Kv1.5-dependent dilation is altered in coronary artery disease, suggesting a role in microvascular dysfunction [132]. Beyond tone regulation, Kv1.5 expression in vascular smooth muscle cells influences phenotypic modulation and proliferative signaling, with oxidative stress further modulating Kv1.5-dependent currents in the microcirculation [133,134]. Prior work from our group showed that co-expression of Kv1.5 with the Kvβ1.2 subunit was associated with a marked reduction in proliferation in HEK293T cells across 24–96 h [52].
Early interest in Kv1.5 centered on its atrial-enriched expression relative to ventricular myocardium and its contribution to atrial repolarization [135] (Figure 1). This atrial-predominant profile supported the early rationale for atrial-selective rhythm control with less ventricular proarrhythmic risk. Subsequent studies identified Kv1.5 expression in VSMC, where K+ efflux influences membrane potential, Ca2+ entry, and vasomotor tone in coronary resistance vessels and the microcirculation [136]. More recent work links Kv1.5 to vascular remodeling through VSMC phenotypic modulation and interaction with Kv1.3 channels [133]. Oxidative stress further modulates Kv1.5-dependent currents, contributing to microvascular dysfunction [134]. At the same time, the clinical translation of selective Kv1.5/ IKur inhibition has been limited by atrial remodeling, reduced target availability in established AF, and the broader, multifactorial substrate that sustains the arrhythmia. In addition, reported KCNA5 variants in Chinese and Scandinavian AF cohorts [30,137] support inter-individual variability in Kv1.5 biology, which may be relevant to precision-targeted therapy. More broadly, the emerging view of Kv-associated channel complexes suggests physiologic relevance beyond atrial electrophysiology alone, raising the possibility that therapies targeting Kv1.5 may have unintended extra-cardiovascular consequences [138]. Thus, the evolving view of Kv1.5 includes not only its atrial electrophysiologic relevance but also its broader extra-atrial safety implications, which may narrow the therapeutic window of Kv1.5-directed therapy in AF.
From a safety standpoint, these vascular roles of Kv1.5 present important considerations for arrhythmia management. Pharmacological inhibition of Kv1.5 for AF could theoretically impair coronary metabolic dilation, reduce myocardial perfusion, or alter microvascular function, effects that would be particularly concerning in patients with underlying coronary artery disease or heart failure. While cardiac therapies often block Kv channels, vascular strategies would generally aim to enhance Kv activity to promote vasodilation [139]. To date, no Kv-specific agent has been approved for vascular indications, and the full extent of vascular off-target effects of Kv1.5 inhibitors remains to be characterized in larger clinical studies.
10. Conclusions
Atrial fibrillation is one of the most rapidly expanding cardiovascular epidemics globally, driven by aging populations, increasing prevalence of cardiometabolic disease, and persistent disparities in detection and treatment. Despite decades of therapeutic innovation, pharmacologic management of AF continues to rely predominantly on antiarrhythmic agents developed in previous decades. These drugs often couple electrophysiologic efficacy with systemic toxicity, increased risk of ventricular proarrhythmia, and the need for intensive monitoring. The strategy of atrial-selective ion channel targeting has emerged as a promising approach to address these limitations. Kv1.5, due to its central role in generating the atrial-specific IKur current, is uniquely positioned to provide safer rhythm-control therapies.
However, the translational experience with Kv1.5 inhibition demonstrates a broader principle in cardiovascular drug development: biological plausibility does not ensure clinical efficacy. Although selective IKur blockade consistently produces atrial electrophysiologic effects and favorable ventricular safety profiles in experimental models, the complex and dynamic substrate of human AF, driven by electrical remodeling, oxidative stress, structural fibrosis, and multi-channel dysregulation, limits the therapeutic benefit of targeting a single ion channel. As atrial fibrillation progresses, the IKur current that initially generated therapeutic optimism becomes downregulated, reducing the effectiveness of Kv1.5 inhibition and highlighting the multifactorial mechanisms underlying arrhythmia maintenance.
Concurrently, recent findings indicate that Kv1.5 biology extends beyond atrial electrophysiology. Kv1.5 participates in broader signaling networks, including redox regulation, mitochondrial function, vascular tone, and coronary microcirculation. This positions Kv1.5 not only as an antiarrhythmic target but also as a component of a larger cardiometabolic regulatory system. These insights broaden the scientific perspective from a narrowly defined electrophysiologic target to a multidimensional biological pathway with implications for vascular disease, myocardial energetics, and tissue perfusion.
The pursuit of safer rhythm-control strategies has implications that extend beyond pharmacology. Many current antiarrhythmic therapies require specialized monitoring or infrastructure, which limits their accessibility in rural areas, resource-limited health systems, and underserved populations where the burden of arrhythmia is increasing most rapidly. Therefore, the ongoing development of atrial-selective or mechanism-based therapies represents both a scientific challenge and a public health priority. This effort aligns with global initiatives to reduce cardiovascular disparities and improve equitable access to arrhythmia care. Emerging evidence of KCNA5 genetic variation across ethnic populations suggests that responses to Kv1.5-targeted therapy may differ between patient groups, highlighting the need for future studies examining how population-level differences in channel biology influence treatment outcomes.
Future therapeutic strategies are expected to move beyond single-channel inhibition toward integrated approaches that address the broader arrhythmogenic substrate. These approaches may combine targeted electrophysiologic modulation with interventions targeting oxidative stress, structural remodeling, and metabolic dysregulation. Within this evolving framework, Kv1.5 serves as an instructive model, illustrating that the progression from molecular discovery to clinical application is rarely straightforward. Each advance in understanding cardiac ion channel biology creates new opportunities to develop safer and more effective arrhythmia therapies.
In summary, the case of Kv1.5 highlights a central principle in contemporary cardiovascular medicine: addressing the challenge of atrial fibrillation will require not only improved pharmacologic agents but also a deeper integration of molecular biology, clinical electrophysiology, and population health. The future of rhythm control may depend not on abandoning atrial-selective targets, but on redefining their role within a broader systems-level understanding of arrhythmia pathophysiology.
Author Contributions
Conceptualization, N.S.Z.; Methodology, N.S.Z., F.M.M. and A.M.O.; Investigation, N.S.Z., F.M.M., A.M.O. and O.A.A.; Writing—Original Draft Preparation, F.M.M., A.M.O., O.A.A. and N.S.Z.; Writing—Review & Editing, F.M.M., A.M.O. and N.S.Z.; Visualization, F.M.M., A.M.O. and N.S.Z.; Supervision, N.S.Z. All authors have read and agreed to the published version of the manuscript.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Funding
This research received no external funding.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
Chugh SS, Havmoeller R, Narayanan K, Singh D, Rienstra M, Benjamin EJ, et al. Worldwide epidemiology of atrial fibrillation: A Global Burden of Disease 2010 Study. Circulation 2014, 129, 837–847. DOI:10.1161/CIRCULATIONAHA.113.005119 [Google Scholar]
-
Linz D, Gawalko M, Betz K, Hendriks JM, Lip GYH, Vinter N, et al. Atrial fibrillation: Epidemiology, screening and digital health. Lancet Reg. Health Eur. 2024, 37, 100786. DOI:10.1016/j.lanepe.2023.100786 [Google Scholar]
-
Lloyd-Jones DM, Wang TJ, Leip EP, Larson MG, Levy D, Vasan RS, et al. Lifetime Risk for Development of Atrial Fibrillation. Circulation 2004, 110, 1042–1046. DOI:10.1161/01.CIR.0000140263.20897.42 [Google Scholar]
-
Vinter N, Cordsen P, Johnsen SP, Staerk L, Benjamin EJ, Frost L, et al. Temporal trends in lifetime risks of atrial fibrillation and its complications between 2000 and 2022: Danish, nationwide, population based cohort study. BMJ 2024, 385, e077209. DOI:10.1136/bmj-2023-077209 [Google Scholar]
-
Essien UR, Kornej J, Johnson AE, Schulson LB, Benjamin EJ, Magnani JW. Social determinants of atrial fibrillation. Nat. Rev. Cardiol. 2021, 18, 763–773. DOI:10.1038/s41569-021-00561-0 [Google Scholar]
-
Bordignon S, Chiara Corti M, Bilato C. Atrial Fibrillation Associated with Heart Failure, Stroke and Mortality. J. Atr. Fibrillation 2012, 5, 467. DOI:10.4022/jafib.467 [Google Scholar]
-
Tamirisa KP, Al-Khatib SM, Mohanty S, Han JK, Natale A, Gupta D, et al. Racial and Ethnic Differences in the Management of Atrial Fibrillation. CJC Open 2021, 3, S137–S148. DOI:10.1016/j.cjco.2021.09.004 [Google Scholar]
-
Bansal N, Xie D, Tao K, Chen J, Deo R, Horwitz E, et al. Atrial Fibrillation and Risk of ESRD in Adults with CKD. Clin. J. Am. Soc. Nephrol. 2016, 11, 1189–1196. DOI:10.2215/CJN.10921015 [Google Scholar]
-
Chao TF, Joung B, Takahashi Y, Lim TW, Choi EK, Chan YH, et al. 2021 Focused Update Consensus Guidelines of the Asia Pacific Heart Rhythm Society on Stroke Prevention in Atrial Fibrillation: Executive Summary. Thromb. Haemost. 2022, 122, 20–47. DOI:10.1055/s-0041-1739411 [Google Scholar]
-
Thompson L, Kurz M. Just Missing the Mark: Discharging High-risk Atrial Fibrillation/Flutter without Thromboprophylaxis. West. J. Emerg. Med. 2018, 19, 361–363. DOI:10.5811/westjem.2018.2.37337 [Google Scholar]
-
Deshmukh A, Iglesias M, Khanna R, Beaulieu T. Healthcare utilization and costs associated with a diagnosis of incident atrial fibrillation. Heart Rhythm O2 2022, 3, 577–586. DOI:10.1016/j.hroo.2022.07.010 [Google Scholar]
-
Kim MH, Johnston SS, Chu BC, Dalal MR, Schulman KL. Estimation of Total Incremental Health Care Costs in Patients with Atrial Fibrillation in the United States. Circ. Cardiovasc. Qual. Outcomes 2011, 4, 313–320. DOI:10.1161/CIRCOUTCOMES.110.958165 [Google Scholar]
-
Li X, Li Z, He H, Wang S, Su H, Kang G. Global burden and health inequality of atrial fibrillation/atrial flutter from 1990 to 2021. Front. Cardiovasc. Med. 2025, 12, 1585980. DOI:10.3389/fcvm.2025.1585980 [Google Scholar]
-
Rani S, Kumar L, Ali SME, Ashraf S, Bhimani S, Kumar S, et al. Trends in United States mortality among patients with atrial fibrillation/flutter related heart failure (1999–2024): Disparities by gender, race/ethnicity and region. BMC Cardiovasc. Disord. 2025, 25, 558. DOI:10.1186/s12872-025-05036-5 [Google Scholar]
-
Martin SS, Aday AW, Allen NB, Almarzooq ZI, Anderson CAM, Arora P, et al. 2025 Heart Disease and Stroke Statistics: A Report of US and Global Data From the American Heart Association. Circulation 2025, 151, e41–e660. DOI:10.1161/CIR.0000000000001303 [Google Scholar]
-
O’Neal WT, Judd SE, Limdi NA, McIntyre WF, Kleindorfer DO, Cushman M, et al. Differential Impact of Risk Factors in Blacks and Whites in the Development of Atrial Fibrillation: The Reasons for Geographic and Racial Differences in Stroke (REGARDS) Study. J. Racial Ethn. Health Disparities 2017, 4, 718–724. DOI:10.1007/s40615-016-0275-3 [Google Scholar]
-
Dewland TA, Olgin JE, Vittinghoff E, Marcus GM. Incident Atrial Fibrillation Among Asians, Hispanics, Blacks, and Whites. Circulation 2013, 128, 2470–2477. DOI:10.1161/CIRCULATIONAHA.113.002449 [Google Scholar]
-
Houmsse A, Malhotra N, Smith SA, El Refaey M. Atrial fibrillation in Black American patients: A review of genetics, risk factors, and outcomes. Heart Rhythm 2025, 22, 617–626. DOI:10.1016/j.hrthm.2024.10.074 [Google Scholar]
-
Stamos TD, Darbar D. The “Double” Paradox of Atrial Fibrillation in Black Individuals. JAMA Cardiol. 2016, 1, 377–379. DOI:10.1001/jamacardio.2016.1259 [Google Scholar]
-
Heckbert SR, Austin TR, Jensen PN, Chen LY, Post WS, Floyd JS, et al. Differences by Race/Ethnicity in the Prevalence of Clinically Detected and Monitor-Detected Atrial Fibrillation. Circ. Arrhythmia Electrophysiol. 2020, 13, e007698. DOI:10.1161/CIRCEP.119.007698 [Google Scholar]
-
Calvert P, Tamirisa K, Al-Ahmad A, Lip GYH, Gupta D. Racial and Ethnic Disparities in Stroke Prevention for Atrial Fibrillation. Am. J. Med. 2023, 136, 225–233. DOI:10.1016/j.amjmed.2022.11.009 [Google Scholar]
-
Wing SL, Jeyakumar N, Kang Y, Stirling K, Wald R, Harel Z. Incidence and Prevalence of Atrial Fibrillation Among Individuals with Chronic Kidney Disease. Kidney Med. 2026, 8, 101184. DOI:10.1016/j.xkme.2025.101184 [Google Scholar]
-
Bansal N, Fan D, Hsu CY, Ordonez JD, Go AS. Incident atrial fibrillation and risk of death in adults with chronic kidney disease. J. Am. Heart Assoc. 2014, 3, e001303. DOI:10.1161/JAHA.114.001303 [Google Scholar]
-
Bautista J, Bella A, Chaudhari A, Pekler G, Sapra KJ, Carbajal R, et al. Advanced chronic kidney disease in non-valvular atrial fibrillation: Extending the utility of R2CHADS2 to patients with advanced renal failure. Clin. Kidney J. 2015, 8, 226–231. DOI:10.1093/ckj/sfv006 [Google Scholar]
-
Parsons C, Cha S, Shen WK, Chamberlain AM, Luis SA, Keddis M, et al. Usefulness of the Addition of Renal Function to the CHA2DS2-VASc Score as a Predictor of Thromboembolism and Mortality in Patients Without Atrial Fibrillation. Am. J. Cardiol. 2018, 122, 597–603. DOI:10.1016/j.amjcard.2018.04.049 [Google Scholar]
-
Mahendru D, Joshi HD, Kowey P. Dofetilide in atrial fibrillation: A comprehensive review. Indian Pacing Electrophysiol. J. 2025, 25, 451–457. DOI:10.1016/j.ipej.2025.11.013 [Google Scholar]
-
Mubarik A, Patel P, Cassagnol M. Sotalol; StatPearls Publishing: Treasure Island, FL, USA, 2026. [Google Scholar]
-
Baumer U, Pedarnig L, Hammer A, Steinacher E, Kazem N, Koller L, et al. The Prognostic Interplay of Heart Failure and Chronic Kidney Disease in Atrial Fibrillation: Focus on Cardiorenal Outcomes. Cardiorenal Med. 2025, 15, 607–618. DOI:10.1159/000548380 [Google Scholar]
-
Kumar A, Kumar C, Kumar A, Kumari S, Aneela, Rai R, et al. Management of anticoagulation in patients with atrial fibrillation and renal dysfunction: A systematic review. World J. Exp. Med. 2025, 15, 105485. DOI:10.5493/wjem.v15.i3.105485 [Google Scholar]
-
Yang Y, Li J, Lin X, Yang Y, Hong K, Wang L, et al. Novel KCNA5 loss-of-function mutations responsible for atrial fibrillation. J. Hum. Genet. 2009, 54, 277–283. DOI:10.1038/jhg.2009.26 [Google Scholar]
-
Ackerman MJ, Tester DJ, Jones GS, Will ML, Burrow CR, Curran ME. Ethnic Differences in Cardiac Potassium Channel Variants: Implications for Genetic Susceptibility to Sudden Cardiac Death and Genetic Testing for Congenital Long QT Syndrome. Mayo Clin. Proc. 2003, 78, 1479–1487. DOI:10.4065/78.12.1479 [Google Scholar]
-
Takimoto K, Levitan ES. Glucocorticoid induction of Kv1.5 K+ channel gene expression in ventricle of rat heart. Circ. Res. 1994, 75, 1006–1013. DOI:10.1161/01.RES.75.6.1006 [Google Scholar]
-
Jeevaratnam K, Chadda KR, Huang CLH, Camm AJC. Cardiac potassium channels: Physiological insights for targeted therapy. J. Cardiovasc. Pharmacol. Ther. 2018, 23, 119–129. DOI:10.1177/1074248417729880 [Google Scholar]
-
Wu W, Sanguinetti MC. Molecular basis of cardiac delayed rectifier K+ channel function and pharmacology. Card. Electrophysiol. Clin. 2016, 8, 275–284. DOI:10.1016/j.ccep.2016.01.002 [Google Scholar]
-
Dong C, Li J, Ding W, Ueda R, Xie X, Wu J, et al. Open channel block of Kv1.5 channels by HMQ1611. Front. Pharmacol. 2022, 13, 965086. DOI:10.3389/fphar.2022.965086 [Google Scholar]
-
Ravens U, Odening KE. Atrial fibrillation: Therapeutic potential of atrial K+ channel blockers. Pharmacol. Ther. 2017, 176, 13–21. DOI:10.1016/j.pharmthera.2016.10.003 [Google Scholar]
-
Lamothe SM, Hogan-Cann AE, Li W, Guo J, Yang T, Tschirhart JN, et al. The N terminus and transmembrane segment S1 of Kv1.5 can coassemble with the rest of the channel independently of the S1–S2 linkage. J. Biol. Chem. 2018, 293, 15347–15358. DOI:10.1074/jbc.RA118.004065 [Google Scholar]
-
Raph SM, Bhatnagar A, Nystoriak MA. Biochemical and physiological properties of K+ channel-associated AKR6A (Kvβ) proteins. Chem. Biol. Interact. 2019, 305, 21–27. DOI:10.1016/j.cbi.2019.03.023 [Google Scholar]
-
McCauley M, Vallabhajosyula S, Darbar D. Proarrhythmic and torsadogenic effects of potassium channel blockers in patients. Card. Electrophysiol. Clin. 2016, 8, 481–493. DOI:10.1016/j.ccep.2016.02.009 [Google Scholar]
-
Wettwer E, Hála O, Christ T, Heubach JF, Dobrev D, Knaut M, et al. Role of IKur in controlling action potential shape and contractility in the human atrium: Influence of chronic atrial fibrillation. Circulation 2004, 110, 2299–2306. DOI:10.1161/01.CIR.0000145155.60288.71 [Google Scholar]
-
Ford JW, Milnes JT. New drugs targeting the cardiac ultra-rapid delayed-rectifier current (IKur): Rationale, pharmacology and evidence for potential therapeutic value. J. Cardiovasc. Pharmacol. 2008, 52, 105–120. DOI:10.1097/FJC.0b013e3181719b0c [Google Scholar]
-
Wirth KJ, Paehler T, Rosenstein B, Knobloch K, Maier T, Frenzel J, et al. Atrial effects of the novel K+-channel-blocker AVE0118 in anesthetized pigs. Cardiovasc. Res. 2003, 60, 298–306. DOI:10.1016/S0008-6363(03)00543-1 [Google Scholar]
-
Voigt N, Dobrev D. Atrial-selective potassium channel blockers. Card. Electrophysiol. Clin. 2016, 8, 411–421. DOI:10.1016/j.ccep.2016.02.005 [Google Scholar]
-
Brown RA, Lau YC, Lip GYH. Vernakalant hydrochloride to treat atrial fibrillation. Expert Opin. Pharmacother. 2014, 15, 865–872. DOI:10.1517/14656566.2014.898751 [Google Scholar]
-
Tamargo J, Caballero R, Gómez R, Delpón E. IKur/Kv1.5 channel blockers for the treatment of atrial fibrillation. Expert Opin. Investig. Drugs 2009, 18, 399–416. DOI:10.1517/13543780902762850 [Google Scholar]
-
Du Y, Wang T, Guo J, Li W, Yang T, Szendrey M, et al. Kv1.5 channels are regulated by PKC-mediated endocytic degradation. J. Biol. Chem. 2021, 296, 100514. DOI:10.1016/j.jbc.2021.100514 [Google Scholar]
-
Ramos-Mondragón R, Lozhkin A, Vendrov AE, Runge MS, Isom LL, Madamanchi NR. NADPH oxidases and oxidative stress in the pathogenesis of atrial fibrillation. Antioxidants 2023, 12, 1833. DOI:10.3390/antiox12101833 [Google Scholar]
-
Su X, Shao B, Chen Z, Gu H, Xiong K, Wang G, et al. SNAP25-dependent membrane trafficking of the Kv1.5 channel regulates the onset of atrial fibrillation. Nat. Commun. 2025, 16, 3730. DOI:10.1038/s41467-025-59096-4 [Google Scholar]
-
Campomanes CR, Carroll KI, Manganas LN, Hershberger ME, Gong B, Antonucci DE, et al. Kv beta subunit oxidoreductase activity and Kv1 potassium channel trafficking. J. Biol. Chem. 2002, 277, 8298–8305. DOI:10.1074/jbc.M110276200 [Google Scholar]
-
Dwenger MM, Raph SM, Baba SP, Moore JB, Nystoriak MA. Diversification of potassium currents in excitable cells via Kvβ proteins. Cells 2022, 11, 2230. DOI:10.3390/cells11142230 [Google Scholar]
-
Tipparaju SM, Li X-P, Kilfoil PJ, Xue B, Uversky VN, Bhatnagar A, et al. Interactions between the C-terminus of Kv1.5 and Kvβ regulate pyridine nucleotide-dependent changes in channel gating. Pflugers Arch. 2012, 463, 799–818. DOI:10.1007/s00424-012-1093-z [Google Scholar]
-
Penzkowski S, Borchardt A, Zhang D, Zinkevich N. Generation and proliferation assessment of HEK293T cells stably expressing Kv1.5 channel with and without regulatory subunits β1.1 or β1.2. FASEB J. 2021, 35. DOI:10.1096/fasebj.2021.35.S1.00204 [Google Scholar]
-
Svoboda LK, Reddie KG, Zhang L, Vesely ED, Williams ES, Schumacher SM, et al. Redox-sensitive sulfenic acid modification regulates surface expression of the cardiovascular voltage-gated potassium channel Kv1.5. Circ. Res. 2012, 111, 842–853. DOI:10.1161/CIRCRESAHA.111.263525 [Google Scholar]
-
Caouette D, Dongmo C, Bérubé J, Fournier D, Daleau P. Hydrogen peroxide modulates the Kv1.5 channel expressed in a mammalian cell line. Naunyn Schmiedeberg’s Arch. Pharmacol. 2003, 368, 479–486. DOI:10.1007/s00210-003-0834-0 [Google Scholar]
-
Li D, Ding Z, Du K, Ye X, Cheng S. Reactive oxygen species as a link between antioxidant pathways and autophagy. Oxid. Med. Cell Longev. 2021, 2021, 5583215. DOI:10.1155/2021/5583215 [Google Scholar]
-
Carnes CA, Chung MK, Nakayama T, Nakayama H, Baliga RS, Piao S, et al. Ascorbate attenuates atrial pacing-induced peroxynitrite formation and electrical remodeling and decreases the incidence of postoperative atrial fibrillation. Circ. Res. 2001, 89, 487–494. DOI:10.1161/hh1801.097644 [Google Scholar]
-
Lu G, Xu C, Tang K, Zhang J, Li Q, Peng L, et al. H2S inhibits angiotensin II-induced atrial Kv1.5 upregulation by attenuating Nox4-mediated ROS generation during atrial fibrillation. Biochem. Biophys. Res. Commun. 2017, 483, 534–540. DOI:10.1016/j.bbrc.2016.12.110 [Google Scholar]
-
Lu G, Xu S, Peng L, Huang Z, Wang Y, Gao X. Angiotensin II upregulates Kv1.5 expression through ROS-dependent transforming growth factor-beta1 and extracellular signal-regulated kinase 1/2 signalings in neonatal rat atrial myocytes. Biochem. Biophys. Res. Commun. 2014, 454, 410–416. DOI:10.1016/j.bbrc.2014.10.088 [Google Scholar]
-
Zhang J, Youn JY, Kim AY, Ramirez RJ, Gao L, Ngo D, et al. NOX4-dependent hydrogen peroxide overproduction in human atrial fibrillation and HL-1 atrial cells: Relationship to hypertension. Front. Physiol. 2012, 3, 140. DOI:10.3389/fphys.2012.00140 [Google Scholar]
-
Dzeshka MS, Shantsila A, Shantsila E, Lip GYH. Atrial fibrillation and hypertension. Hypertension 2017, 70, 854–861. DOI:10.1161/HYPERTENSIONAHA.117.08934 [Google Scholar]
-
Bonilla IM, Sridhar A, Györke S, Cardounel AJ, Carnes CA. Nitric oxide synthases and atrial fibrillation. Front. Physiol. 2012, 3, 105. DOI:10.3389/fphys.2012.00105 [Google Scholar]
-
Dudley SC, Jr., Hoch NE, McCann LA, Honeycutt C, Diamandopoulos L, Fukai T, et al. Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: Role of the NADPH and xanthine oxidases. Circulation 2005, 112, 1266–1273. DOI:10.1161/CIRCULATIONAHA.105.538108 [Google Scholar]
-
Simon JN, Ziberna K, Casadei B. Compromised redox homeostasis, altered nitroso–redox balance, and therapeutic possibilities in atrial fibrillation. Cardiovasc. Res. 2016, 109, 510–518. DOI:10.1093/cvr/cvw012 [Google Scholar]
-
Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JGN, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: A mitochondria-ROS-HIF-1α-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H570–H578. DOI:10.1152/ajpheart.01324.2007 [Google Scholar]
-
Lagrutta A, Wang J, Fermini B, Salata JJ. Novel, potent inhibitors of human Kv1.5 K+ channels and ultrarapidly activating delayed rectifier potassium current. J. Pharmacol. Exp. Ther. 2006, 317, 1054–1063. DOI:10.1124/jpet.106.101162 [Google Scholar]
-
Ravens U, Wettwer E. Ultra-rapid delayed rectifier channels: Molecular basis and therapeutic implications. Cardiovasc. Res. 2011, 89, 776–785. DOI:10.1093/cvr/cvq398 [Google Scholar]
-
Ehrlich J, Biliczki P, Hohnloser S, Nattel S. Atrial-selective approaches for the treatment of atrial fibrillation. J. Am. Coll. Cardiol. 2008, 51, 787–792. DOI:10.1016/j.jacc.2007.08.067 [Google Scholar]
-
Regan CP, Wallace AA, Cresswell HK, Atkins CL, Lynch JJ, Jr. In vivo cardiac electrophysiologic effects of a novel diphenylphosphine oxide IKur blocker, (2-isopropyl-5-methylcyclohexyl) diphenylphosphine oxide, in rat and nonhuman primate. J. Pharmacol. Exp. Ther. 2006, 316, 727–732. DOI:10.1124/jpet.105.094839 [Google Scholar]
-
Regan CP, Kiss L, Stump GL, McIntyre CJ, Beshore DC, Liverton NJ, et al. Atrial antifibrillatory effects of structurally distinct IKur blockers 3-[(dimethylamino)methyl]-6-methoxy-2-methyl-4-phenylisoquinolin-1(2 H)-one and 2-phenyl-1,1-dipyridin-3-yl-2-pyrrolidin-1-yl-ethanol in dogs with underlying heart failure. J. Pharmacol. Exp. Ther. 2008, 324, 322–330. DOI:10.1124/jpet.107.127654 [Google Scholar]
-
Borrego J, Feher A, Jost N, Panyi G, Varga Z, Papp F. Peptide inhibitors of Kv1.5: An option for the treatment of atrial fibrillation. Pharmaceuticals 2021, 14, 1303. DOI:10.3390/ph14121303 [Google Scholar]
-
Pavri BB, Greenberg HE, Kraft WK, Lazarus N, Lynch JJ, Salata JJ, et al. MK-0448, a specific Kv1.5 inhibitor: Safety, pharmacokinetics, and pharmacodynamic electrophysiology in experimental animal models and humans. Circ. Arrhythm. Electrophysiol. 2012, 5, 1193–1201. DOI:10.1161/CIRCEP.111.969782 [Google Scholar]
-
Loose S, Mueller J, Wettwer E, Knaut M, Ford J, Milnes J, et al. Effects of IKur blocker MK-0448 on human right atrial action potentials from patients in sinus rhythm and in permanent atrial fibrillation. Front. Pharmacol. 2014, 5, 26. DOI:10.3389/fphar.2014.00026 [Google Scholar]
-
Blandin CE, Gravez BJ, Hatem SN, Balse E. Remodeling of ion channel trafficking and cardiac arrhythmias. Cells 2021, 10, 2417. DOI:10.3390/cells10092417 [Google Scholar]
-
Van Wagoner DR, Pond AL, McCarthy PM, Trimmer JS, Nerbonne JM. Outward K+ current densities and Kv1.5 expression are reduced in chronic human atrial fibrillation. Circ. Res. 1997, 80, 772–781. DOI:10.1161/01.RES.80.6.772 [Google Scholar]
-
Ford J, Milnes J, Wettwer E, Christ T, Rogers M, Sutton K, et al. Human electrophysiological and pharmacological properties of XEN-D0101: A novel atrial-selective Kv1.5/IKur inhibitor. J. Cardiovasc. Pharmacol. 2013, 61, 408–415. DOI:10.1097/FJC.0b013e31828780eb [Google Scholar]
-
Tsiachris D, Kotoulas SC, Doundoulakis I, Antoniou C, Botis M, Pamporis K, et al. Reappraising Use of Flecainide for Atrial Fibrillation and Ventricular Arrhythmias in Structural Heart Disease Patients. Medicina 2025, 61, 1845. DOI:10.3390/medicina61101845 [Google Scholar]
-
Florek JB, Lucas A, Girzadas D. Amiodarone; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
-
Bardy GH, Lee KL, Mark DB, Poole JE, Packer DL, Boineau R, et al. Amiodarone or an implantable cardioverter-defibrillator for congestive heart failure. N. Engl. J. Med. 2005, 352, 225–237. DOI:10.1056/NEJMoa043399 [Google Scholar]
-
Naccarelli GV, Wolbrette DL, Levin V, Samii S, Banchs JE, Penny-Peterson E, et al. Safety and efficacy of dronedarone in the treatment of atrial fibrillation/flutter. Clin. Med. Insights Cardiol. 2011, 5, CMC.S6677. DOI:10.4137/CMC.S6677 [Google Scholar]
-
Hohnloser SH, Crijns HJGM, van Eickels M, Gaudin C, Page RL, Torp-Pedersen C, et al. Effect of Dronedarone on Cardiovascular Events in Atrial Fibrillation. N. Engl. J. Med. 2009, 360, 668–678. DOI:10.1056/NEJMoa0803778 [Google Scholar]
-
Køber L, Torp-Pedersen C, McMurray JJV, Gøtzsche O, Lévy S, Crijns H, et al. Increased Mortality after Dronedarone Therapy for Severe Heart Failure. N. Engl. J. Med. 2008, 358, 2678–2687. DOI:10.1056/NEJMoa0800456 [Google Scholar]
-
Flaker GC. A Clinical Trial on Trial: The Results of PALLAS. American College of Cardiology. Available online: https://www.acc.org/latest-in-cardiology/articles/2014/07/18/15/02/a-clinical-trial-on-trial-the-results-of-pallas (accessed on 4 February 2026).
-
Le Heuzey J, De Ferrari GM, Radzik D, Santini M, Zhu J, Davy JM. A Short-Term, Randomized, Double-Blind, Parallel-Group Study to Evaluate the Efficacy and Safety of Dronedarone versus Amiodarone in Patients with Persistent Atrial Fibrillation: The DIONYSOS Study. J. Cardiovasc. Electrophysiol. 2010, 21, 597–605. DOI:10.1111/j.1540-8167.2010.01764.x [Google Scholar]
-
Brophy JM, Nadeau L. Amiodarone versus dronedarone for atrial fibrillation—A retrospective cohort study. CJC Open 2022, 5, 8–14. DOI:10.1016/j.cjco.2022.09.008 [Google Scholar]
-
Roden DM. Cellular basis of drug-induced torsades de pointes. Br. J. Pharmacol. 2008, 154, 1502–1507. DOI:10.1038/bjp.2008.238 [Google Scholar]
-
Waldo AL, Camm AJ, deRuyter H, Friedman PL, MacNeil DJ, Pauls JF, et al. Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. Lancet 1996, 348, 7–12. DOI:10.1016/S0140-6736(96)02149-6 [Google Scholar]
-
Danish Investigations of Arrhythmia and Mortality on Dofetilide. American College of Cardiology. Available online: https://www.acc.org/Latest-in-Cardiology/Clinical-Trials/2010/02/23/19/01/DIAMOND (accessed on 3 February 2026).
-
Pedersen OD, Bagger H, Keller N, Marchant B, Køber L, Torp-Pedersen C, et al. Efficacy of dofetilide in the treatment of atrial fibrillation-flutter in patients with reduced left ventricular function: A Danish Investigations of Arrhythmia and Mortality ON Dofetilide (DIAMOND) substudy. Circulation 2001, 104, 292–296. DOI:10.1161/01.CIR.104.3.292 [Google Scholar]
-
Ellenbogen KA, Clemo HF, Stambler BS, Wood MA, VanderLugt JT. Efficacy of ibutilide for termination of atrial fibrillation and flutter. Am. J. Cardiol. 1996, 78, 42–45. DOI:10.1016/S0002-9149(96)00565-6 [Google Scholar]
-
Nowak RM, Bodnar TJ, Dronen S, Gentzkow G, Tomlanovich MC. Bretylium tosylate as initial treatment for cardiopulmonary arrest: Randomized comparison with placebo. Ann. Emerg. Med. 1981, 10, 404–407. DOI:10.1016/S0196-0644(81)80306-X [Google Scholar]
-
Marinelli A, Ciccarelli I, Capucci A. Vernakalant hydrochloride in the treatment of atrial fibrillation: A review of the latest clinical evidence. Clin. Investig. 2011, 1, 579–588. DOI:10.4155/cli.11.27 [Google Scholar]
-
Akel T, Lafferty J. Efficacy and safety of intravenous vernakalant for the rapid conversion of recent-onset atrial fibrillation: A meta-analysis. Ann. Noninvasive Electrocardiol. 2018, 23, e12508. DOI:10.1111/anec.12508 [Google Scholar]
-
Bacaner MB. Quantitative comparison of bretylium with other antifibrillatory drugs. Am. J. Cardiol. 1968, 21, 504–512. DOI:10.1016/0002-9149(68)90282-8 [Google Scholar]
-
Singh BN. Amiodarone: The expanding antiarrhythmic role and how to follow a patient on chronic therapy. Clin. Cardiol. 1997, 20, 608–618. DOI:10.1002/clc.4960200706 [Google Scholar]
-
Szymanski MW, Pellegrini MV, Cassagnol M. Ibutilide; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
-
Camm AJ, Capucci A, Hohnloser SH, Torp-Pedersen C, Van Gelder IC, Mangal B, et al. A randomized active-controlled study comparing the efficacy and safety of vernakalant to amiodarone in recent-onset atrial fibrillation. J. Am. Coll. Cardiol. 2011, 57, 313–321. DOI:10.1016/j.jacc.2010.07.046 [Google Scholar]
-
Kowey PR, Dorian P, Mitchell LB, Pratt CM, Roy D, Schwartz PJ, et al. Vernakalant hydrochloride for the rapid conversion of atrial fibrillation after cardiac surgery: A randomized, double-blind, placebo-controlled trial. Circ. Arrhythm. Electrophysiol. 2009, 2, 652–659. DOI:10.1161/CIRCEP.109.870204 [Google Scholar]
-
Camm J. The vernakalant story: How did it come to approval in Europe and what is the delay in the U.S.A.? Curr. Cardiol. Rev. 2014, 10, 309–314. DOI:10.2174/1573403X10666140513103709 [Google Scholar]
-
Ford J, Milnes J, El Haou S, Wettwer E, Loose S, Matschke K, et al. The positive frequency-dependent electrophysiological effects of the IKur inhibitor XEN-D0103 are desirable for the treatment of atrial fibrillation. Heart Rhythm. 2016, 13, 555–564. DOI:10.1016/j.hrthm.2015.10.003 [Google Scholar]
-
Comes N, Bielanska J, Vallejo-Gracia A, Serrano-Albarrás A, Marruecos L, Gómez D, et al. The voltage-dependent K+ channels Kv1.3 and Kv1.5 in human cancer. Front. Physiol. 2013, 4, 283. DOI:10.3389/fphys.2013.00283 [Google Scholar]
-
Blaauw Y, Schotten U, van Hunnik A, Neuberger HR, Allessie MA. Cardioversion of persistent atrial fibrillation by a combination of atrial specific and non-specific class III drugs in the goat. Cardiovasc. Res. 2007, 75, 89–98. DOI:10.1016/j.cardiores.2007.03.021 [Google Scholar]
-
Cardiovascular Pharmacology Concepts. Class III Antiarrhythmics (Potassium Channel Blockers). Available online: https://cvpharmacology.com/antiarrhythmic/class-III (accessed on 25 March 2026).
-
Riera ARP, Uchida AH, Ferreira C, Ferreira Filho C, Schapachnik E, Dubner S, et al. Relationship among amiodarone, new class III antiarrhythmics, miscellaneous agents and acquired long QT syndrome. Cardiol. J. 2008, 15, 209–219. Available online: https://journals.viamedica.pl/cardiology_journal/article/view/21599 (accessed on 25 March 2026).
-
Jaiswal A, Goldbarg S. Dofetilide induced torsade de pointes: Mechanism, risk factors and management strategies. Indian Heart J. 2014, 66, 640–648. DOI:10.1016/j.ihj.2013.12.021 [Google Scholar]
-
Gowda RM, Khan IA, Punukollu G, Vasavada BC, Sacchi TJ, Wilbur SL. Female preponderance in ibutilide-induced torsade de pointes. Int. J. Cardiol. 2004, 95, 219–222. DOI:10.1016/j.ijcard.2003.04.034 [Google Scholar]
-
Wongsalap Y, Miliam W, Deesham S, Thepsaen A, Churasae A, Poolpun D, et al. Incidence and risk factors of QT prolongation and torsades de pointes following intravenous amiodarone administration for atrial fibrillation: A cohort study. Arch. Acad. Emerg. Med. 2025, 13, e70. DOI:10.22037/aaem.v13i1.2784 [Google Scholar]
-
Anderson JL. Bretylium tosylate: Profile of the only available class III antiarrhythmic agent. Clin. Ther. 1985, 7, 205–224. Available online: https://europepmc.org/article/med/3886143 (accessed on 25 March 2026).
-
Bretylium Side Effects. Available online: https://www.drugs.com/sfx/bretylium-side-effects.html (accessed on 25 March 2026).
-
Wolkove N, Baltzan M. Amiodarone pulmonary toxicity. Can. Respir. J. 2009, 16, 282540. DOI:10.1155/2009/282540 [Google Scholar]
-
Vassallo P, Trohman RG. Prescribing amiodarone: An evidence-based review of clinical indications. JAMA 2007, 298, 1312–1322. DOI:10.1001/jama.298.11.1312 [Google Scholar]
-
Dofetilide (Oral Route). Available online: https://www.mayoclinic.org/drugs-supplements/dofetilide-oral-route/description/drg-20063516 (accessed on 25 March 2026).
-
Glatter K, Yang Y, Chatterjee K, Modin G, Cheng J, Kayser S, et al. Chemical cardioversion of atrial fibrillation or flutter with ibutilide in patients receiving amiodarone therapy. Circulation 2001, 103, 253–257. DOI:10.1161/01.CIR.103.2.253 [Google Scholar]
-
Ragucci KR. Elevated liver enzymes associated with dronedarone for atrial fibrillation. SAGE Open Med. Case Rep. 2013, 1, 2050313X13511600. DOI:10.1177/2050313X13511600 [Google Scholar]
-
Schweizer P, Becker R, Katus HA, Thomas D. Dronedarone: Current evidence for its safety and efficacy in the management of atrial fibrillation. Drug Des. Devel Ther. 2011, 5, 27–39. DOI:10.2147/DDDT.S10315 [Google Scholar]
-
Wesley RC, Jr., Farkhani F, Morgan D, Zimmerman D. Ibutilide: Enhanced defibrillation via plateau sodium current activation. Am. J. Physiol. Heart Circ. Physiol. 1993, 264, H1269–H1274. DOI:10.1152/ajpheart.1993.264.4.H1269 [Google Scholar]
-
Bryan CK, Darby MH. Bretylium tosylate: A review. Am. J. Hosp. Pharm. 1979, 36, 1189–1192. DOI:10.1093/ajhp/36.9.1189 [Google Scholar]
-
Bretylium (Rx). Available online: https://reference.medscape.com/drug/bretylium-1000321 (accessed on 25 March 2026).
-
Chen Y, Liu Z, Li Y, Wang W, Chen T, Li S, et al. Drug–drug interactions and combination therapy strategies of amiodarone with digoxin, rivaroxaban, and phenytoin assessed by physiologically based pharmacokinetic modeling. Pharmacotherapy 2025, 45, 566–577. DOI:10.1002/phar.70050 [Google Scholar]
-
11 Amiodarone Interactions: Antibiotics, Statins, and More. Available online: https://www.goodrx.com/amiodarone/interactions (accessed on 25 March 2026).
-
Ibrahim MA, Tivakaran VS. Dofetilide; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
-
Calvo D, Filgueiras-Rama D, Jalife J. Mechanisms and Drug Development in Atrial Fibrillation. Pharmacol. Rev. 2018, 70, 505–525. DOI:10.1124/pr.117.014183 [Google Scholar]
-
Priest BT, McDermott JS. Cardiac ion channels. Channels 2015, 9, 352–359. DOI:10.1080/19336950.2015.1076597 [Google Scholar]
-
Podd SJ, Freemantle N, Furniss SS, Sulke N. First clinical trial of specific IKAChblocker shows no reduction in atrial fibrillation burden in patients with paroxysmal atrial fibrillation: Pacemaker assessment of BMS 914392 in patients with paroxysmal atrial fibrillation. Europace 2015, 18, 340–346. DOI:10.1093/europace/euv263 [Google Scholar]
-
Mhanna M, Al-Abdouh A, Alzahrani A, Jabri A, Al-Harbi A, Beran A, et al. Decades of Shifting Trends: Arrhythmia-Related Mortalities in the United States (1968–2021) with 2040 Projections. J. Am. Heart Assoc. 2025, 14, e039615. DOI:10.1161/JAHA.124.039615 [Google Scholar]
-
Treewaree S, Lip GYH. An Updated Global Perspective of Atrial Fibrillation: Trends, Risk Factors, and Socioeconomic Disparities. CJC Open 2025, 7, 259–261. DOI:10.1016/j.cjco.2024.12.003 [Google Scholar]
-
Sharma SP, Turagam M, Atkins D, Bommana S, Jeffrey C, Newton D, et al. Safety of rapid switching from amiodarone to dofetilide in atrial fibrillation patients with an implantable cardioverter–defibrillator. Heart Rhythm. 2019, 16, 990–995. DOI:10.1016/j.hrthm.2019.01.028 [Google Scholar]
-
American College of Cardiology. Dofetilide (Tikosyn) Considerations for Use. Available online: https://www.acc.org/~/media/Non-Clinical/Files-PDFs-Excel-MS-Word-etc/Tools%20and%20Practice%20Support/Quality%20and%20Clinical%20Toolkits/AFib%20Toolkit/Dofetilide.pdf?la=en (accessed on 4 February 2026).
-
U.S. Food and Drug Administration. Information for Tikosyn (Dofetilide). Available online: https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/information-tikosyn-dofetilide (accessed on 4 February 2026).
-
U.S. Food and Drug Administration. Tikosyn (Dofetilide) Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/020931s017lbl.pdf (accessed on 4 February 2026).
-
Berg T. Kv7(KCNQ)-K+-Channels Influence Total Peripheral Resistance in Female but Not Male Rats, and Hamper Catecholamine Release in Hypertensive Rats of Both Sexes. Front. Physiol. 2018, 9, 117. DOI:10.3389/fphys.2018.00117 [Google Scholar]
-
Ohanyan V, Yin L, Bardakjian R, Kolz C, Enrick M, Hakobyan T, et al. Requisite role of Kv1.5 channels in coronary metabolic dilation. Circ. Res. 2015, 117, 612–621. DOI:10.1161/CIRCRESAHA.115.306642 [Google Scholar]
-
Nishijima Y, Cao S, Chabowski DS, Korishettar A, Ge A, Zheng X, et al. Contribution of KV1.5 channel to hydrogen peroxide–induced human arteriolar dilation and its modulation by coronary artery disease. Circ. Res. 2017, 120, 658–669. DOI:10.1161/CIRCRESAHA.116.309491 [Google Scholar]
-
Zhang DX, Gutterman DD. Myocardin and Kv1 channels: A paradigm shift in treating vascular smooth muscle cell–related proliferative disease? Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2454–2456. DOI:10.1161/ATVBAHA.119.313531 [Google Scholar]
-
Chen WL, Huang XQ, Zhao LY, Li J, Chen JW, Xiao Y, et al. Involvement of Kv1.5 protein in oxidative vascular endothelial cell injury. PLoS ONE 2012, 7, e49758. DOI:10.1371/journal.pone.0049758 [Google Scholar]
-
Fedida D, Eldstrom J, Hesketh JC, Lamorgese M, Castel L, Steele DF, et al. Kv1.5 is an important component of repolarizing K+ current in canine atrial myocytes. Circ. Res. 2003, 93, 744–751. DOI:10.1161/01.RES.0000096362.60730.AE [Google Scholar]
-
Jackson WF. KV channels and the regulation of vascular smooth muscle tone. Microcirculation 2018, 25, e12421. DOI:10.1111/micc.12421 [Google Scholar]
-
Christophersen IE, Olesen MS, Liang B, Andersen MN, Larsen AP, Nielsen JB, et al. Genetic variation in KCNA5: Impact on the atrial-specific potassium current IKur in patients with lone atrial fibrillation. Eur. Heart J. 2013, 34, 1517–1525. DOI:10.1093/eurheartj/ehs442 [Google Scholar]
-
Mohamed ZA, Li J, Wen J, Jia F, Banerjee S. The KCNB2 gene and its role in neurodevelopmental disorders: Implications for genetics and therapeutic advances. Clin. Chim. Acta 2025, 566, 120056. DOI:10.1016/j.cca.2024.120056 [Google Scholar]
-
Zhao Z, Ruan S, Ma X, Feng Q, Xie Z, Nie Z, et al. Challenges Faced with Small Molecular Modulators of Potassium Current Channel Isoform Kv1.5. Biomolecules 2020, 10, 10. DOI:10.3390/biom10010010 [Google Scholar]