Found 26 results
Open Access
Article
24 June 2024Sulforaphane’s Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2)-Dependent and -Independent Mechanism of Anti-SARS-CoV-2 Activity
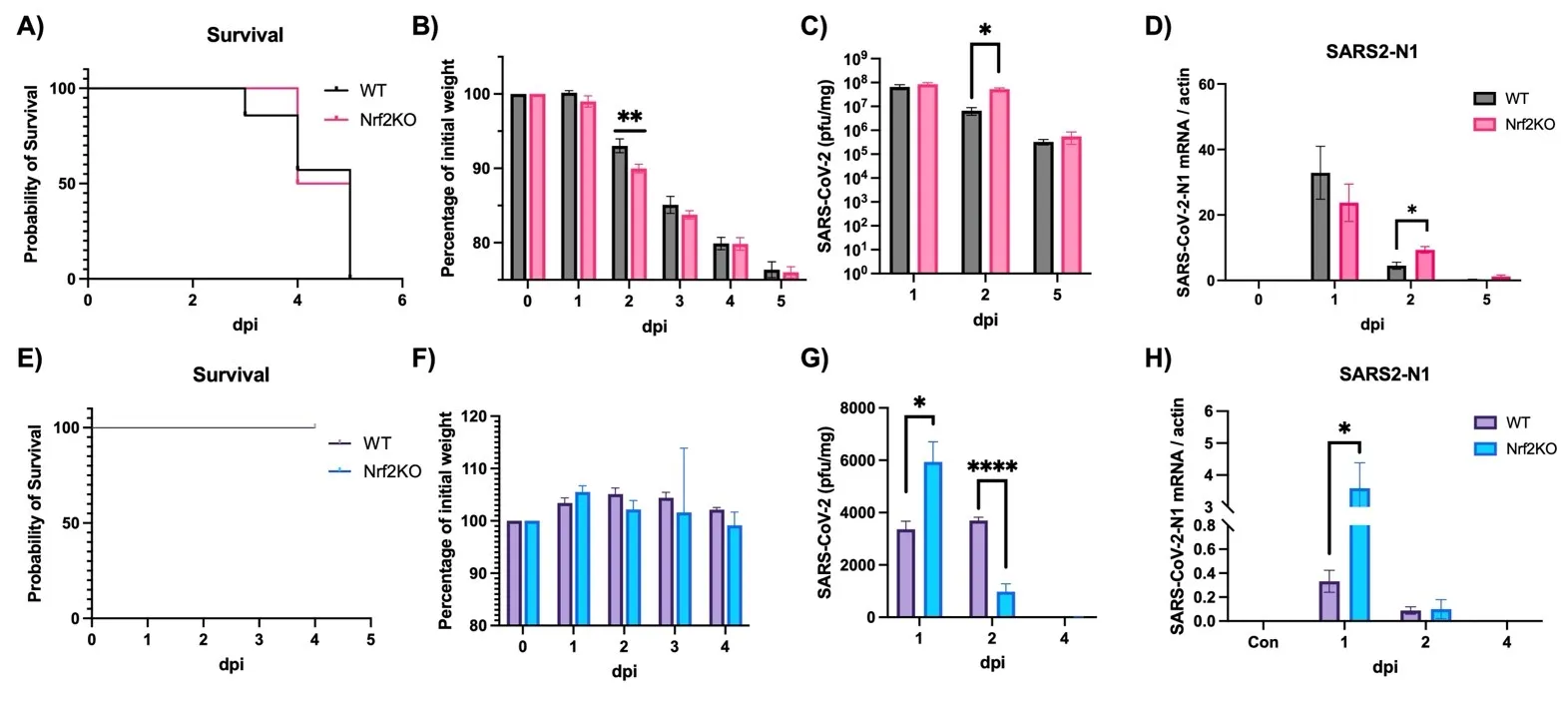
It is well established that Nrf2 plays a crucial role in anti-oxidant and anti-inflammatory functions. However, its antiviral capabilities remain less explored. Despite this, several Nrf2 activators have demonstrated anti-SARS-CoV-2 properties, though the mechanisms behind these effects are not fully understood. In this study, using two mouse models of SARS-CoV-2 infection, we observed that the absence of Nrf2 significantly increased viral load and altered inflammatory responses. Additionally, we evaluated five Nrf2 modulators. Notably, epigallocatechin gallate (EGCG), sulforaphane (SFN), and dimethyl fumarate (DMF) exhibited significant antiviral effects, with SFN being the most effective. SFN did not impact viral entry but appeared to inhibit the main protease (MPro) of SARS-CoV-2, encoded by the Nsp5 gene, as indicated by two protease inhibition assays. Moreover, using two Nrf2 knockout cell lines, we confirmed that SFN's antiviral activity occurs independently of Nrf2 activation in vitro. Paradoxically, in vivo tests using the MA30 model showed that SFN's antiviral function was completely lost in Nrf2 knockout mice. Thus, although SFN and potentially other Nrf2 modulators can inhibit SARS-CoV-2 independently of Nrf2 activation in cell models, their Nrf2-dependent activities might be crucial for antiviral defense under physiological conditions.
Open Access
Commentary
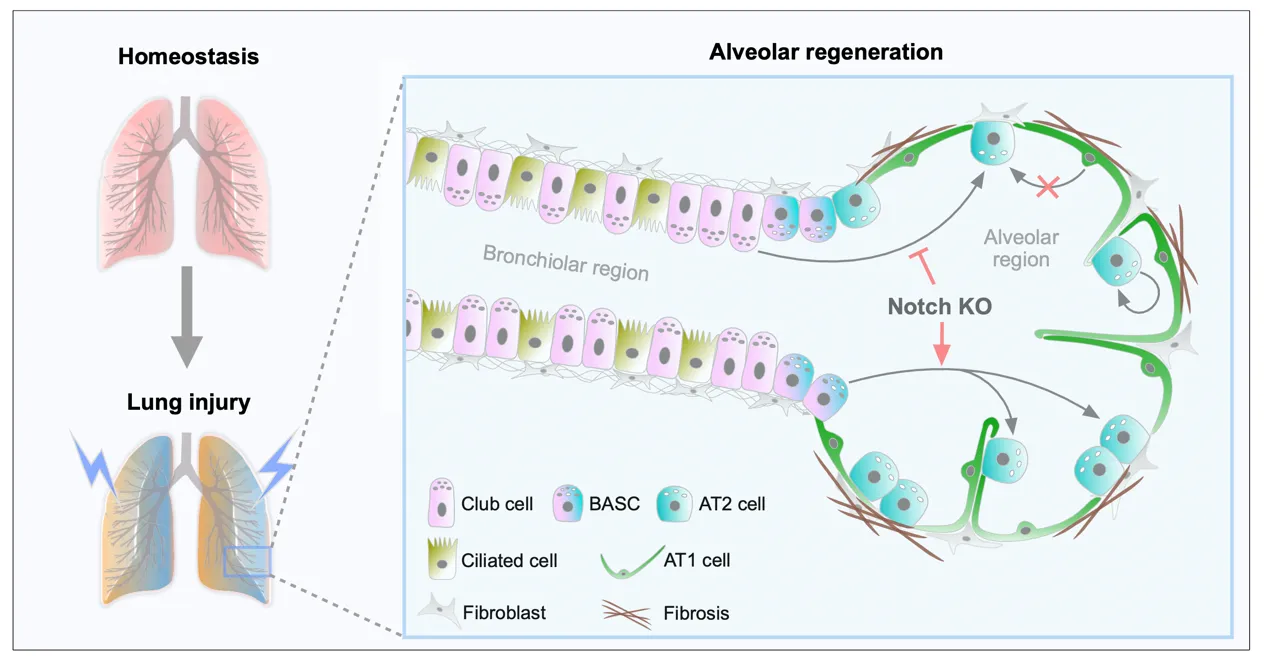
12 June 2024Dual Genetic Tracing Reveals the Origin of Alveolar Stem Cells after Lung Injury
As alveolar epithelial stem cells, alveolar type II (AT2) cells play a pivotal role in sustaining alveolar homeostasis and facilitating repair processes. However, the sources of AT2 cell regeneration have remained contentious due to the non-specific labeling limitations of traditional single recombinase-based lineage tracing techniques. To address this issue, we employed dual recombination systems to develop more precise lineage tracing methodologies, effectively bypassing the shortcomings of conventional approaches and enabling specific labeling of lung epithelial cells. Our findings demonstrate that, following lung injury, regenerated AT2 cells do not originate from alveolar type I (AT1) cells, but instead derive from bronchiolar club cells and bronchioalveolar stem cells (BASCs), alongside the self-renewal of resident AT2 cells. Furthermore, we discovered that the transition of club cells and BASCs into AT2 cells is distinctly modulated by the Notch signaling pathway. This study not only provides novel insights into lung regeneration, but the innovative lineage tracing technology developed herein also holds promise as a technical support for research in diverse fields.
Open Access
Communication
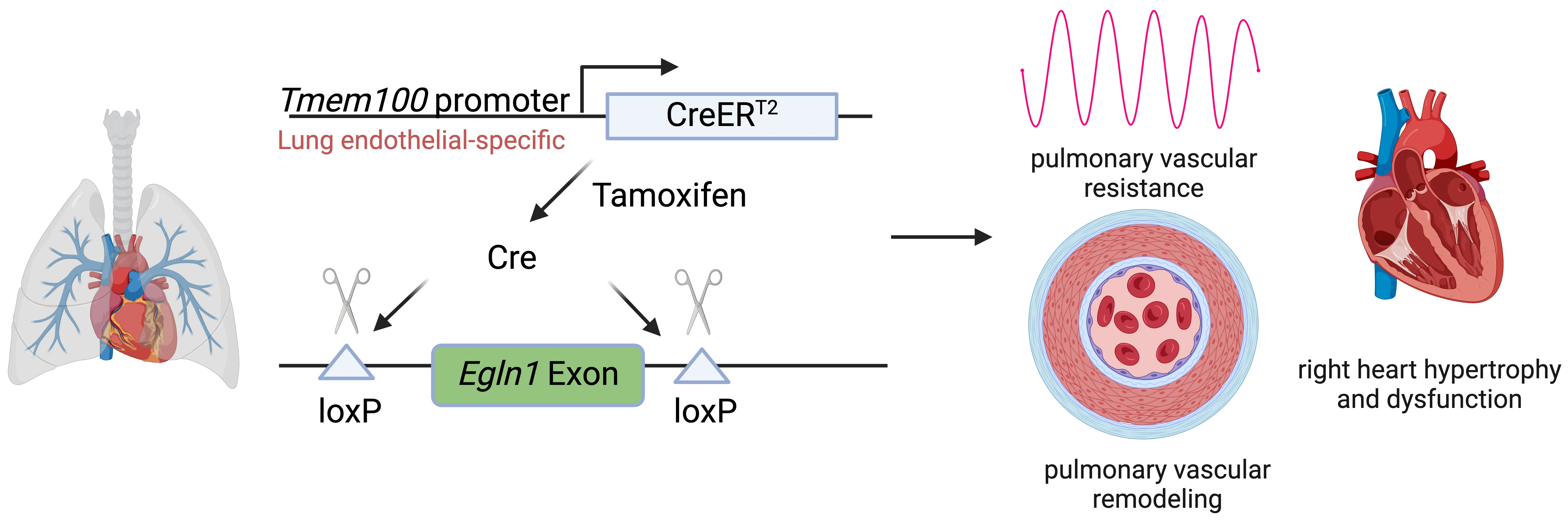
30 May 2024A Novel Animal Model for Pulmonary Hypertension: Lung Endothelial-Specific Deletion of Egln1 in Mice
Pulmonary arterial hypertension (PAH) is a devastating disease characterized by high blood pressure in the pulmonary arteries, which can potentially lead to heart failure over time. Previously, our lab found that endothelia-specific knockout of Egln1, encoding prolyl 4-hydroxylase-2 (PHD2), induced spontaneous pulmonary hypertension (PH). Recently, we elucidated that Tmem100 is a lung-specific endothelial gene using Tmem100-CreERT2 mice. We hypothesize that lung endothelial-specific deletion of Egln1 could lead to the development of PH without affecting Egln1 gene expression in other organs. Tmem100-CreERT2 mice were crossed with Egln1flox/flox mice to generate Egln1f/f;Tmem100-CreERT2 (LiCKO) mice. Western blot and immunofluorescent staining were performed to verify the knockout efficacy of Egln1 in multiple organs of LiCKO mice. PH phenotypes, including hemodynamics, right heart size and function, pulmonary vascular remodeling, were evaluated by right heart catheterization and echocardiography measurements. Tamoxifen treatment induced Egln1 deletion in the lung endothelial cells (ECs) but not in other organs of adult LiCKO mice. LiCKO mice exhibited an increase in right ventricular systolic pressure (RVSP, ~35 mmHg) and right heart hypertrophy. Echocardiography measurements showed right heart hypertrophy, as well as cardiac and pulmonary arterial dysfunction. Pulmonary vascular remodeling, including increased pulmonary wall thickness and muscularization of distal pulmonary arterials, was enhanced in LiCKO mice compared to wild-type mice. Tmem100 promoter-mediated lung endothelial knockout of Egln1 in mice leads to development of spontaneous PH. LiCKO mice could serve as a novel mouse model for PH to study lung and other organ crosstalk.
Open Access
Article
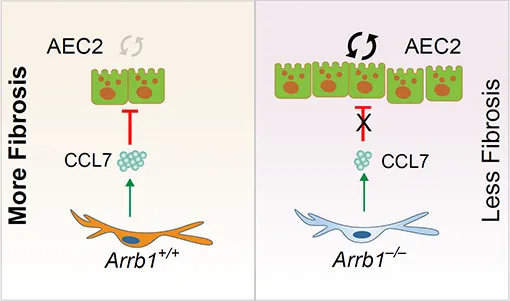
30 April 2024Arrestin beta 1 Regulates Alveolar Progenitor Renewal and Lung Fibrosis
The molecular mechanisms that regulate progressive pulmonary fibrosis remain poorly understood. Type 2 alveolar epithelial cells (AEC2s) function as adult stem cells in the lung. We previously showed that there is a loss of AEC2s and a failure of AEC2 renewal in the lungs of idiopathic pulmonary fibrosis (IPF) patients. We also reported that beta-arrestins are the key regulators of fibroblast invasion, and beta-arrestin 1 and 2 deficient mice exhibit decreased mortality, decreased matrix deposition, and increased lung function in bleomycin-induced lung fibrosis. However, the role of beta-arrestins in AEC2 regeneration is unclear. In this study, we investigated the role and mechanism of Arrestin beta 1 (ARRB1) in AEC2 renewal and in lung fibrosis. We used conventional deletion as well as cell type-specific deletion of ARRB1 in mice and found that Arrb1 deficiency in fibroblasts protects mice from lung fibrosis, and the knockout mice exhibit enhanced AEC2 regeneration in vivo, suggesting a role of fibroblast-derived ARRB1 in AEC2 renewal. We further found that Arrb1-deficient fibroblasts promotes AEC2 renewal in 3D organoid assays. Mechanistically, we found that CCL7 is among the top downregulated cytokines in Arrb1 deficient fibroblasts and CCL7 inhibits AEC2 regeneration in 3D organoid experiments. Therefore, fibroblast ARRB1 mediates AEC2 renewal, possibly by releasing chemokine CCL7, leading to fibrosis in the lung.
Open Access
Article
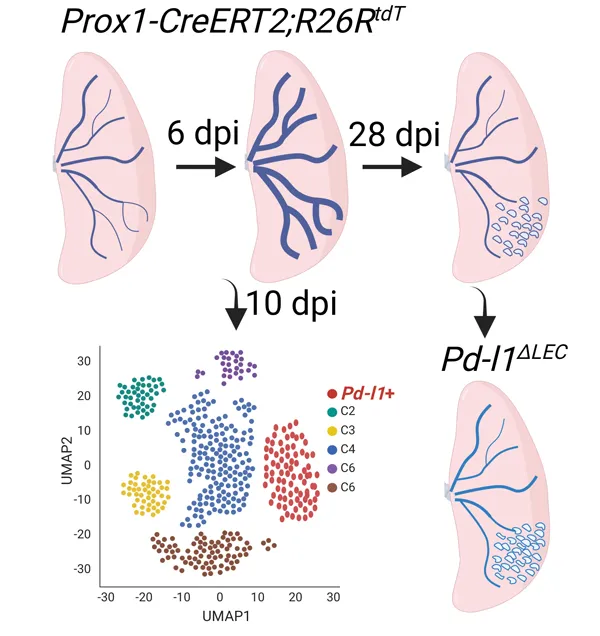
19 February 2024Single Cell Analysis of Lung Lymphatic Endothelial Cells and Lymphatic Responses during Influenza Infection
Tissue lymphatic vessels network plays critical roles in immune surveillance and tissue homeostasis in response to pathogen invasion, but how lymphatic system per se is remolded during infection is less understood. Here, we observed that influenza infection induces a significant increase of lymphatic vessel numbers in the lung, accompanied with extensive proliferation of lymphatic endothelial cells (LECs). Single-cell RNA sequencing illustrated the heterogeneity of LECs, identifying a novel PD-L1+ subpopulation that is present during viral infection but not at steady state. Specific deletion of Pd-l1 in LECs elevated the expansion of lymphatic vessel numbers during viral infection. Together these findings elucidate a dramatic expansion of lung lymphatic network in response to viral infection, and reveal a PD-L1+ LEC subpopulation that potentially modulates lymphatic vessel remolding.
Open Access
Article
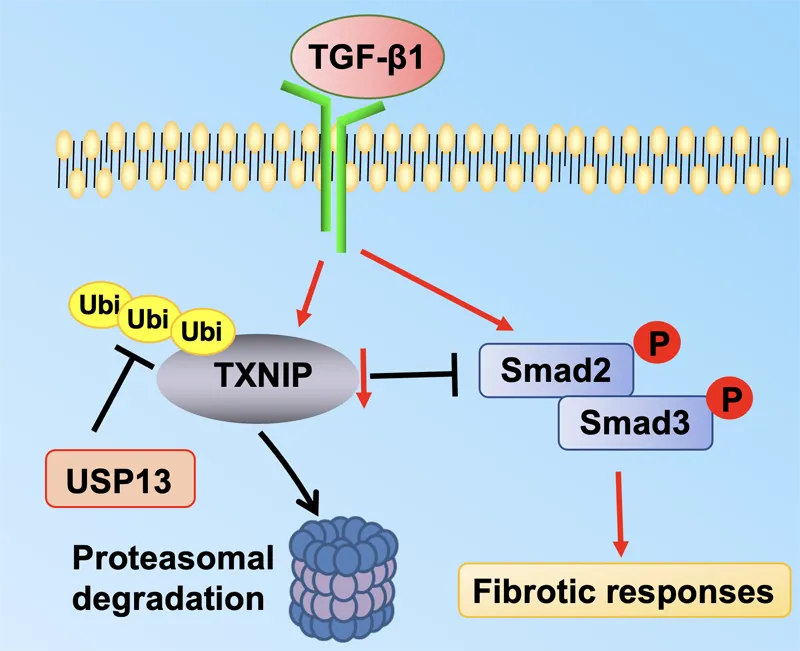
01 February 2024Molecular Regulation of Transforming Growth Factor-β1-induced Thioredoxin-interacting Protein Ubiquitination and Proteasomal Degradation in Lung Fibroblasts: Implication in Pulmonary Fibrosis
Thioredoxin-interacting protein (TXNIP) plays a critical role in regulation of cellular redox reactions and inflammatory responses by interacting with thioredoxin (TRX) or the inflammasome. The role of TXNIP in lung fibrosis and molecular regulation of its stability have not been well studied. Therefore, here we investigated the molecular regulation of TXNIP stability and its role in TGF-β1-mediated signaling in lung fibroblasts. TXNIP protein levels were significantly decreased in lung tissues from bleomycin-challenged mice. Overexpression of TXNIP attenuated transforming growth factor-β1 (TGF-β1)-induced phosphorylation of Smad2/3 and fibronectin expression in lung fibroblasts, suggesting that decrease in TXNIP may contribute to the pathogenesis of lung fibrosis. Further, we observed that TGF-β1 lowered TXNIP protein levels, while TXNIP mRNA levels were unaltered by TGF-β1 exposure. TGF-β1 induced TXNIP degradation via the ubiquitin-proteasome system. A serine residue mutant (TNXIP-S308A) was resistant to TGF-β1-induced degradation. Furthermore, downregulation of ubiquitin-specific protease-13 (USP13) promoted the TGF-β1-induced TXNIP ubiquitination and degradation. Mechanistic studies revealed that USP13 targeted and deubiquitinated TXNIP. The results of this study revealed that the decrease of TXNIP in lungs apparently contributes to the pathogenesis of pulmonary fibrosis and that USP13 can target TXNP for deubiquitination and regulate its stability.