Most photochemical reactions occur under UV or visible light. Using resonant excitation in the IR region to initiate various processes in molecular systems is attractive since sharp bands of vibrational transitions enable one to excite selectively vibrations of certain molecules, distinguishing even different isotopomers [
1]. However, studies of the resonant interaction of laser radiation with molecules in gas phase face some difficulties. The activation energies of homogeneous chemical reactions usually exceed the energy of infrared quanta, and multiphoton resonant excitation of molecules to high energies is limited by the anharmonicity, which makes vibrational energy levels not equidistant, and the second quantum absorbed by a vibrationaly excited molecule should not have the same frequency as the first one.
Surface processes have, as a rule, lower activation energy as compared to reactions in gases and are sensitive to variations in the excitation energy of the participating molecules. Absorption of one infrared photon by a molecule can significantly change its behavior, which makes it possible to use relatively low laser radiation powers. It was shown [
2] that vibrational excitation of a certain isotopologue of gaseous CO
2 results in a difference in adsorption properties on a cooled surface compared to unexcited molecules. Recently [
3] showed that resonant vibrational excitation of CF
3Br mixed with argon by a CO
2 laser leads to bromine isotope-selective suppression of clustering.
Direct excitation of vibrational states of adsorbed particles in order to initiate surface processes by resonance laser radiation seems even more promising. There were many works with the attempts to invoke desorption induced by resonant laser excitation, when the excitation causes desorption of those molecules whose vibrational frequency coincides with the frequency of the exciting radiation. These studies’ most important practical aim was to estimate the possibility of using resonant excitation of vibrational states of adsorbed species for isotope separation.
Experiments on laser desorption by resonance excitation of vibrations of adsorbed molecules were carried out with CO [
4,
5,
6], CH
3F [
7], and ammonia [
8,
9]. It was found, however, that exposing the mixture to radiation tuned to the vibrational frequency of a specific isotopologue did not produce its anticipated selective desorption, which likely contributed to the decline of research activity in this area in the following years. Notable examples that can be cited include studies on resonant desorption of N
2O [
10] and CD
3F [
11] from the NaCl surface.
Using the time-of-flight mass spectrometry, Redlich et al. [
12] investigated IR laser-stimulated desorption from a multilayer films of different methane isotopologues deposited on a NaCl (100) single-crystal surface. The study examined desorption from layers of pure CH
4, CD
4, or CD
3H, as well as from mixed layers containing two different isotopic species, as a function of the wavelength of a tunable free-electron infrared laser. Desorption of methane was observed exclusively when the incident radiation was tuned to the resonant frequencies of the internal vibrational modes of the molecules. In the case of mixed isotope layers, desorption occurred solely at resonant frequencies and did not depend on the energy density of the exciting laser. The results demonstrated that vibrational excitation of one isotopologue could cause the desorption of another one.
The above findings indicate that vibrational excitation energy is rapidly exchanged within the adsorbed molecular layer. Clarifying the absence of isotope selectivity requires examining the mechanism and efficiency of this energy exchange, assessing the lifetime and dissipation rate of vibrational excitation, and, as far as possible, the probability of inducing desorption or reaction of vibrationally excited surface species. We performed such an analysis [
13], and based on the results, we identified the criteria for systems in which isotope selectivity could be anticipated.
The vibrational energy dissipation time for the absorbed molecules should be relatively long; therefore, suitable materials are insulators or wide-bandgap semiconductors rather than metals, where excitation of electron–hole pairs causes rapid quenching of vibrational states. To reduce phonon generation, the molecules should be weakly bound to the surface, and the frequency of the excited vibrations should be as far as possible from the phonon mode frequencies. Considering that for most oxide adsorbents the lattice vibrations are close to 1000 cm
−1 or lower, it is preferable to select excitation radiation frequencies of at least 2000 cm
−1. At this frequency, the quantum energy (>25 kJ/mol) is sufficient to desorb weakly bound molecules, to stimulate isomeric transitions in systems with linkage isomerism, or to activate reactions of intermediate surface species stabilized at −196 °C. This value significantly exceeds the average thermal energy, this excludes the spontaneous population of these vibrational states even under ambient conditions.
In adsorbed molecular layers, vibrational energy transfer proceeds via the mechanism of resonant dipole–dipole interaction [
14]. The efficiency of this process is governed by the molecule’s vibrational polarizability, which determines the absorption coefficient of the corresponding mode, as well as by the separation between interacting molecules. Energy transfer can be reduced by selecting molecules with moderate vibrational band intensities and using comparatively low surface coverages.
Furthermore, to avoid reverse reactions, the process should be irreversible. Since it is impossible to satisfy all of the aforementioned requirements within a single system, priority was given to irreversibility, and we have selected ozone as the object of study in our first work [
15]. Dissociation of gaseous ozone as a result of multiphoton excitation by a CO
2 laser was studied by Proch and Schröder [
16]. When adsorbed on certain oxide catalysts, ozone decomposes readily at ambient temperature, while on active cerium oxides this reaction can become explosive even at −196 °С [
17]. IR studies have shown that, on the surface of TiO
2 and CeO
2, ozone molecules are distorted, lose their symmetry [
17,
18], and their vibrational frequencies are noticeably shifted with respect to those of gaseous O
3. It was supposed that this form of adsorption can be an intermediate in the ozone decomposition pathway. For ozone of mixed isotopic composition chemisorbed on TiO
2, the ν
1 + ν
3 combination band in the region of 2100 cm
−1 is split into eight individual peaks, each of them was unambiguously assigned [
19], enabling selective excitation of specific isotopologues bound to titanium atoms in a certain manner.
In the adsorbed state, ozone easily reacts with various molecules: for instance, ozonolysis of ethylene on silica occurs efficiently even at −196 °С [
20]. The reaction proceeds slower for chlorine derivatives of ethylene, but the TiO
2 surface catalyzes the ozonolysis of dichloroethylene. Hydrating the titania surface can reduce the reaction rate, and an attempt was made to enhance it via resonant excitation of vibrational states of adsorbed ozone [
15].
Despite the successful detection of redistribution of the absorption band intensities of adsorbed molecules of different isotopic compositions caused by the resonant excitation of certain isotopic modifications, it should be recognized that the choice of ozone is not optimal for achieving the task. Indeed, the absorption band of the ν
1 + ν
3 combination vibration, at the frequency of which the excitation was carried out, has a low intensity, and the amount of absorbed radiation energy is very small. At the same time, the frequencies of vibrations ν
1 and ν
3 are close to the frequencies of lattice vibrations. This causes a high probability of energy transfer to the lattice, and the high intensity of the bands of these vibrations increases this probability and promotes the process of energy exchange between adsorbed molecules of different isotopic composition.
That is why in our further search we used molecules whose frequencies of fundamental vibrations are high enough, and in this paper we summarize the results of these studies, both successful or not, to show the problems, outline the ways to overcome, and to describe the perspectives of these studies.
3.1. Systems with Linkage Isomerism
The ability of CO for linkage isomerism when the molecule could be attached to the same cation in zeolites either by carbon or by oxygen atom [
22] seems rather promising for initiating the isomeric transitions by resonance laser irradiation. Unfortunately, the barrier between the two states is too low, and even at 196 °C, there is a thermodynamic equilibrium between them. To freeze the spontaneous transitions, the attempt to the isomeric transitions of CO adsorbed on KY zeolite by resonance laser irradiation at the frequency of the overtone vibration of the C-bonded complex was performed at −265 °C (8 K). The energy of exciting quantum at 4320 cm
−1 is about 52 kJ/mole, which is much above the adsorption enthalpy, enough for the molecule to be desorbed and readsorbed. One could, thus, anticipate that with a certain probability, it could be adsorbed via the oxygen atom, and the number of O-bonded species could increase. However, after irradiation at 4320 cm
−1, the intensity of the band at
ca 2122 cm
−1, clearly observable at temperatures above −196 °C, remained negligible.
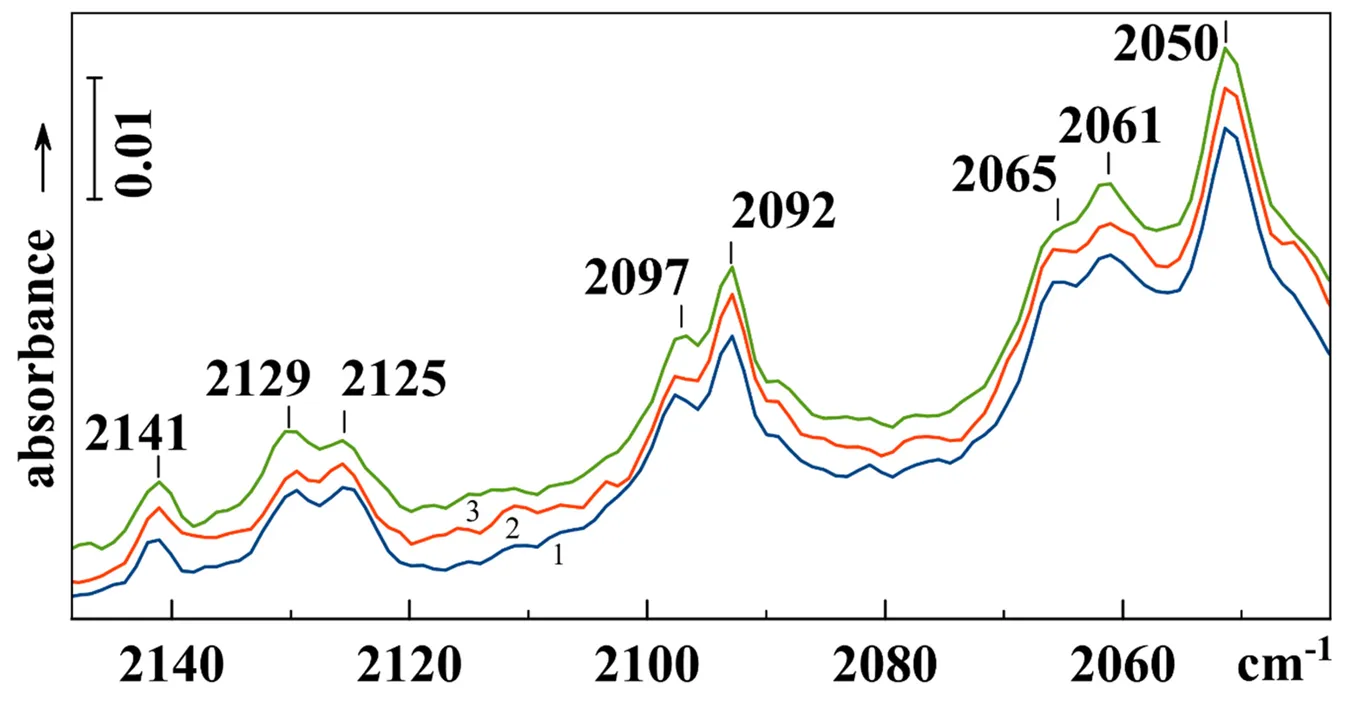
FTIR spectra of CO adsorbed on reduced Ni-containing USY zeolites with Si/Al = 30 reveal formation of mono-, di- and tricarbonyls with Ni
+ sites [
23]. In the spectra of isotopically mixed nickel dicarbonyl species, both the bands are split in two maxima. This phenomenon, called isotopic isomerism, is that the species exists in two isomeric forms, where two CO molecules occupy non-equivalent positions. Then structures with different location of isotopically labeled atom can be distinguished in the spectra. While usual Ni(
12CO)
2 dicarbonyls have two distinct bands at 2141 and 2097 cm
−1, mixed
12CO
13CO dicarbonyl species exhibit two pairs of bands at 2129, 2125 and 2065, 2061 cm
−1. The difference in the adsorption sites cannot explain the splitting since neither band of monocarbonyl, nor those of dicarbonyls of certain isotopic content, exhibit any hint of splitting or even broadening. Quantum mechanical calculation of cluster models of zeolite fragments [
24] has shown that both the molecules are bound to the Ni
+ ion via carbon and that these two CO molecules have different locations in zeolite cages.
The systems with isotopic isomerism seem promising for the experiments with resonant-induced phenomena. Their main advantage, to our mind, is the equal energy of the two states. This prevents spontaneous transitions to the most stable state and makes the mutual transformations between the states equally probable. The nickel dicarbonyls are stable enough and do not desorb up to ambient temperature, thus one could anticipate that at least at −196 °C or even higher, the equilibrium between the two states will not establish itself.
shows the preliminary experiment results with isotopically mixed CO adsorbed on Ni-containing USY zeolite. In accordance with [
23], after removing the excess of adsorbate by pumping in the cell cooled by liquid nitrogen, we observe the bands of dicarbonyls. A pair of bands at 2141 and 2097 cm
−1 are due to Ni(
12CO)
2, those at 2092 and 2050 cm
−1 to Ni(
13CO)
2. The isotopically mixed Ni(
12CO)(
13CO) dicarbonyls account for two doublets at 2129, 2125 and 2065, 2061 cm
−1. As seen from the figure, irradiation at the frequency of the high-frequency band at −196 °C, when the temperature was fixed by the presence of helium, has almost no effect upon the spectrum. However, when irradiation was accompanied by evacuation and the sample temperature was 30–50 °С higher, the resulting intensity redistribution could be clearly seen. Simultaneous relative intensity increase of the bands at 2129 and 2061 cm
−1 confirms their assignment to the same surface species, although the mechanism of a process which could lead to the increase of irradiated compound is not clear.
. FTIR spectrum of <sup>13</sup>CO(55%) adsorbed at −196 °C on the reduced Ni-USY zeolite after pumping in the cooled cell and addition 0.5 Torr of He (<em>1</em>), irradiation at 2129 cm<sup>−1</sup> for 15 min (<em>2</em>) and irradiation at 2129 cm<sup>−1</sup> for 15 min on pumping and addition of He (<em>3</em>).
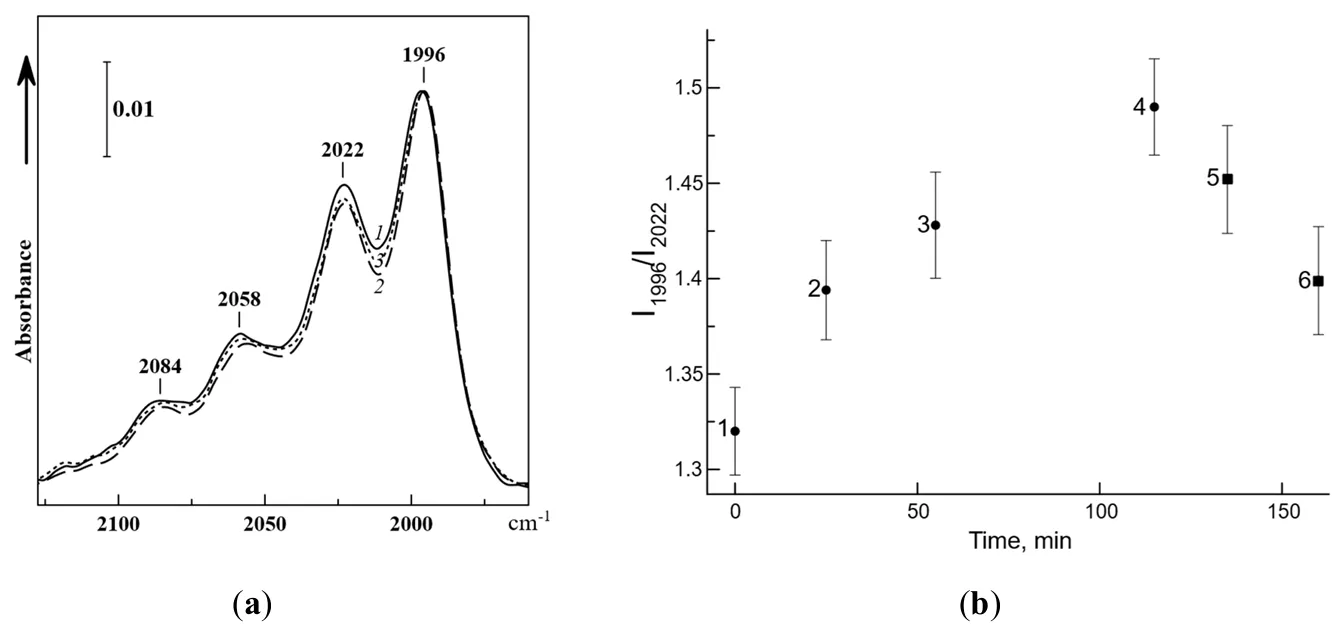
a shows the results of experiment on the influence of IR radiation upon the spectra of ozone adsorbed on TiO
2, partially reported in [
15]. The bands in the figure are due to ozone of mixed isotopic composition (76%
18O) in the region of the combination vibration ν
1 + ν
3, where all curves are normalized to the low-frequency band at 1996 cm
−1 of
18O
3 molecules.
As can be seen, laser irradiation at the frequency of 2030–2020 cm
−1 causes a slight decrease in the intensity of the band at the frequency of laser relative to that at 1996 cm
−1. Meanwhile, in the intervals between sessions, the restoration of the initial intensity ratio between these bands with time is observed. Subsequent irradiation at the frequency of 2200 cm
−1, outside of ozone absorption, accelerates the return to the initial intensity ratio.
b shows the time dependence of the intensity ratio of the 2022 and 1996 cm
−1 bands, determined by decomposing the band contour into components. After several irradiation sessions at 2030–2020 cm
−1, the intensity ratio, which was initially 1.32, grows up to 1.49. Irradiation at 2200 cm
−1 demonstrates the reverse dynamics of intensity changes.
. (<strong>a</strong>) IR spectrum of ozone (76% <sup>18</sup>O) adsorbed at −196 °C on TiO<sub>2</sub> 70 min after insertion (<em>1</em>), after irradiation at a frequency of 2030–2020 cm<sup>−1</sup> for 115 min (<em>2</em>), and subsequent irradiation at 2200 cm<sup>−1</sup> for 45 min (<em>3</em>). The spectra are normalized to the 1996 cm<sup>−1</sup> band; (<strong>b</strong>) Dynamics of changes in the ratio of integral intensities of bands at 1996 and 2022 cm<sup>−1</sup> in the IR spectrum of ozone on irradiation at 2030–2020 cm<sup>−1</sup> for 0 (<em>1</em>), 25 (<em>2</em>), 55 (<em>3</em>) and 115 min (<em>4</em>) and following irradiation at 2200 cm<sup>−1</sup> for 20 (<em>5</em>) and 45 min (<em>6</em>) [<a href="#B15" class="html-bibr">15</a>].
Spectra shown in
were obtained in the presence of weakly bound ozone, which could be removed by prolonged evacuation of the cooled cell, leaving 8 bands of isotopic mixture of chemisorbed O
3, as reported in [
19]. The bands are too weak to follow their intensity changes, but the results of irradiation could be observed in the region of ozone fundamental vibrations. However, no changes were detected after irradiation at the conditions close to those in
. Apparently, the observed effect of irradiation concerned the physisorbed molecules.
Hydroazide acid has certain advantages over ozone for experiments with surface processes caused by resonant laser excitation. It has strong bands of fundamental vibrations in the relatively high-frequency range of about 3300 and 2150 cm
−1, which is quite far from the lattice vibrations of most adsorbents. Thus, the proportion of absorbed light is greater, and the dissipation of vibrational energy by exchanging it for lattice vibrations is less efficient compared to the O
3 molecule. The exchange of vibrational excitation energy in the layer of adsorbed molecules by the resonant dipole–dipole interaction can be reduced by lowering surface coverage without significant loss of the share of absorbed radiation. Nevertheless, our attempt to use laser radiation with the frequency of the 2150 cm
−1 band of HN
3 at −196 °C did not lead to any noticeable changes in the spectrum of HN
3 adsorbed on silica [
25].
3.3. Irradiation-Induced Reactions
One can suppose that vibrationally excited molecules have different activity with respect to coadsorbed molecules. We have tried to find out if the vibrational excitation of ozone accelerates the ozonolysis of
cis-dichloroethylene adsorbed on TiO
2 [
15].
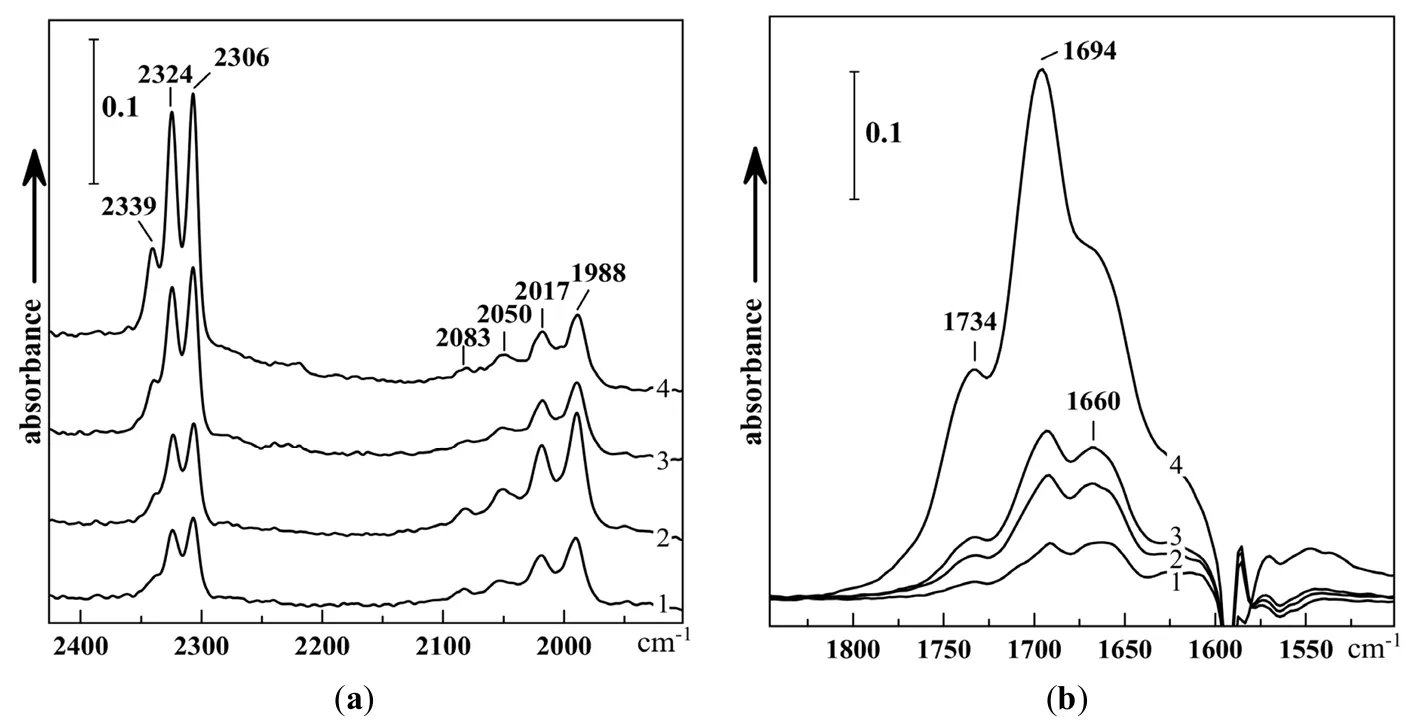
At low temperature when the reaction does not go, irradiation at the frequency of certain isotopic modification was tried. The interaction between adsorbed
cis-dichloroethylene and ozone of mixed isotopic composition produced spectral changes, shown in
. After ozone is inserted, the increase of absorption band at 1694 cm
−1 with shoulders at 1734 and 1660 cm
−1 is observed, which continues with irradiation at the frequency of 2015–2025 cm
−1, when the relative intensity of the shoulder at 1732 cm
−1 slightly increases, while that of the shoulder at 1665 cm
−1, on the contrary, decreases. The same changes occur on irradiation at 2060–2045 cm
−1 and continue with increasing temperature. The intensity ratio of the bands of different CO
2 and ozone isotopologues shown in
enables one to estimate the degree of enrichment of both molecules in
18O. Such an estimate for ozone gives a value close to 76%, while for CO
2, if we base on the intensities of the 2324 and 2306 cm
−1 bands, it turns out to be about 70%.
Experiments on ozonolysis of
cis-dichloroethylene reveal that on TiO
2 the reaction occurs spontaneously, but can be accelerated by irradiation. It results in the formation of a product with a characteristic band at 1694 cm
−1 and a weaker one at 1338 cm
−1 (not shown in
). These bands are close to the frequencies of formyl chloride HCOCl, which, is likely the principal product of the incomplete oxidation of
cis-dichloroethylene on ozonolysis.
. IR spectra of ozonolysis products of <em>cis</em>-C<sub>2</sub>H<sub>2</sub>Cl<sub>2 </sub>adsorbed on TiO<sub>2</sub> in the high-frequency (<strong>a</strong>) and low-frequency (<strong>b</strong>) regions: 40 min after insertion of ozone <sup>18</sup>O<sub>3</sub> (76% <sup>18</sup>O) at −196 °C (<em>1)</em>; after irradiation for 100 min at 2025–2015 cm<sup>−1</sup> (<em>2</em>), irradiation for 70 min more at 2060–2045 cm<sup>−1</sup> (<em>3</em>), and after heating to −100 °C (<em>4</em>). The spectrum of the sample before ozone addition is subtracted [<a href="#B15" class="html-bibr">15</a>].
Complete oxidation leads to the appearance of carbon dioxide, whose weak bands begin to grow after ozone introduction and become stronger upon irradiation. Considering the negligible vapor pressure of CO
2 at −196 °C, it can be inferred that carbon dioxide is generated in situ during ozonolysis rather than introduced as an impurity with ozone.The degree of oxygen enrichment in carbon dioxide determined from the intensity ratio of the components of the split CO
2 band is slightly lower than in the used
18O
3 (76%). This means that nearly all oxygen atoms originate from ozone, and a small decrease in the
18O isotope content may be due to the exchange with surface oxygen.
Attempts to irradiate the isotopically mixed ozone—
cis-dichloroethylene system at various frequencies produced no significant spectral differences. Furthermore, irradiation outside the absorption region of ozone or simply raising the temperature results in comparable changes. If irradiation at the frequency of a specific ozone isotopologue should yield formyl chloride enriched in a particular isotope, confirming this experimentally would be nearly impossible. This experimentally would be nearly impossible. The difficulty arises not only from the broadness of the product bands. Even with normal ozone
16O
3 we observe a complex spectrum of several overlapping bands of products. Bands of isotopically substituted products are close to certain bands of product with the conventional isotope. So, the absorption band of the isotopic analogue of at 1734 cm
−1 band falls at 1697 cm
−1, almost coinciding with the 1694 cm
−1 band of product with usual isotope, and the band of isotopically substituted analogue of the latter at 1665 cm
−1 band is superimposed onto another band at 1660 cm
−1.
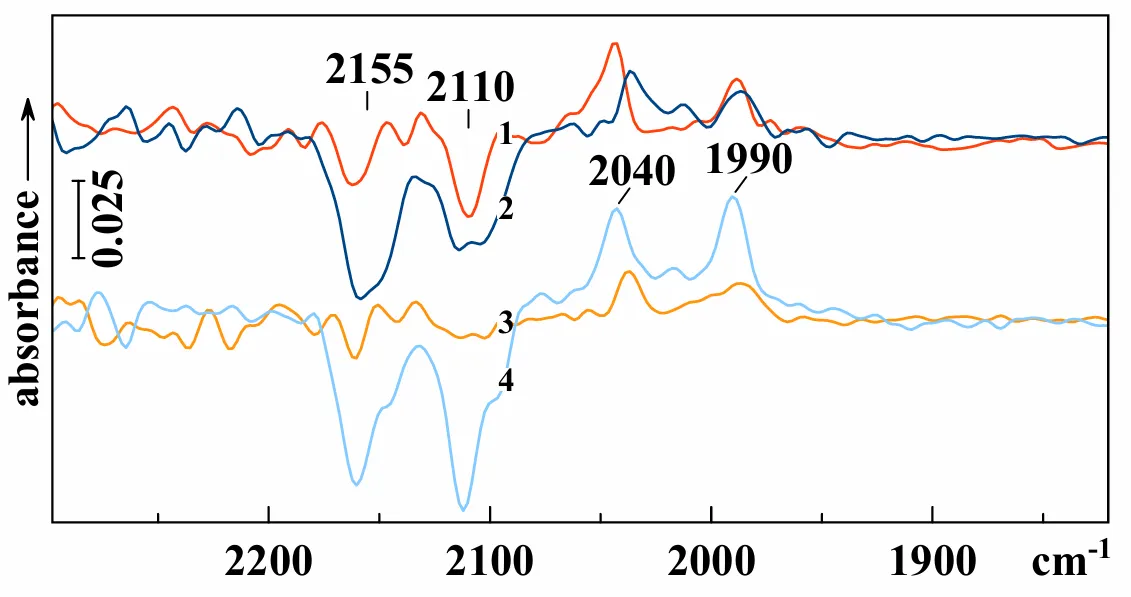
Another surface reaction we tried to stimulate by IR irradiation occurs when CO is adsorbed on CaO prepared by hydroxide decomposition at 700 °C. First, CO molecules on such a surface react with the basic oxygen ions, forming “carbonite” CO
22− ions [
21,
26]. The latter interact with the next CO molecule forming dioxoketene O=C=CO
22− ions with a characteristic band at about 2040 cm
−1 (for
12C
16O). This reaction starts at temperature well above −196 °C and at room temperature is followed by a chain of transformations, including disproportionation resulting in the formation of carbonates and reduced polymeric compounds [
27]. If a small dose of a certain isotopologue of CO is first added at ambient or not very low temperature, and then CO of other isotopic content is added, dioxoketenes of mixed isotopic composition can be obtained. At −196 °C, even at the excess of the added gas evidenced in the spectrum by a band of molecular adsorption at about 2155 cm
−1 (
12C
16O) or 2110 cm
−1 (
13C
16O), dioxoketenes do not arise. We hoped that vibrational excitation of adsorbed CO could provoke the reaction of excited molecules with carbonite ions, and activation of certain isotopologue would lead to the preferable formation of certain isotopic modification of the product.
Preliminary results of such experiments are shown in
. Irradiation at low temperature at the frequency of
13C
16O leads predominantly to the formation of the light (or half-substituted) isotopologue of dioxoketene with a band near 2040 cm
−1, mostly at the expense of the band of
13C
16O at 2110 cm
−1 (curve 1). Raising the temperature results in the growth of both the dioxoketene bands, while the two bands of molecular CO isotopologues diminish simultaneously (curve 4). Although the mechanism of the reaction should be studied in more detail, the result shows that the action of IR irradiation cannot be explained by local heating of the sample.
. Changes in the FTIR spectrum of CaO pretreated at 700 °С with preliminary adsorbed dose of <sup>12</sup>CO after addition of <sup>13</sup>CO (55%) at −196 °C and irradiation at −150 °С for 60 min at 2100–2113 cm<sup>−1</sup> (<em>1</em>) and after heating up to −100 °С for 7 min (<em>2</em>); re-activated sample as in (<em>1</em>), but irradiated at −160 °С for 17 min at 2100–2113 cm<sup>−1</sup> (<em>3</em>) and heated up to −113 °С for 5 min (<em>4</em>). Each curve shows the difference between the spectra taken before and after the mentioned time interval.
There are no cations on the surface of SiO
2 aerosil, and the only adsorption site are silanol groups. Their acidic properties in hydrogen bonding with pre-adsorbed base molecules, such as 2,6-dimethylpyridine (DMP), may be enhanced by interaction of acidic molecules with silanol oxygen atom. Addition of SO
2 leads to the proton transfer from hydroxyl group to the base molecule, revealing the effect of induced Brønsted acidity [
28]. Dimethylpyridine is the most convenient base for studying this phenomenon, since its aromatic ring vibrations ν
8a and ν
8b are very sensitive to the interaction with different surface sites. While in the spectrum of liquid, they are observed at 1594 and 1580 cm
−1, formation of H-bond with silanol groups shifts them to 1606 and 1585 cm
−1, proton transfer to DMP molecule results in their increase up to 1655 and 1629 cm
−1, respectively, outside the absorption region of the neutral molecules. This enables us to use HN
3 adsorption on a sample with pre-adsorbed DMP to evaluate the acidic properties of the acid. It was found that HN
3 addition results in protonation of a part of H-bonded DMP, and we decided to investigate the possibility of accelerating the process of DMP protonation by the vibrational excitation of adsorbed HN
3 molecules.
Adsorption of HN
3 on aerosil, besides the perturbation of silanol groups and appearance of the bands of adsorbed molecules at 3290, 2150, and 1290 cm
−1, close to the frequencies of gaseous acid, leads to a band at 2050 cm
−1, particularly intense for samples with pre−adsorbed DMP and assigned to N
3− ion [
25]. A band at almost the same position (2044–2041 cm
−1) appears in the spectrum of reduced CeO
2 after NO adsorption. Its assignment to the N
3− ion was confirmed by isotopic substitution [
29].
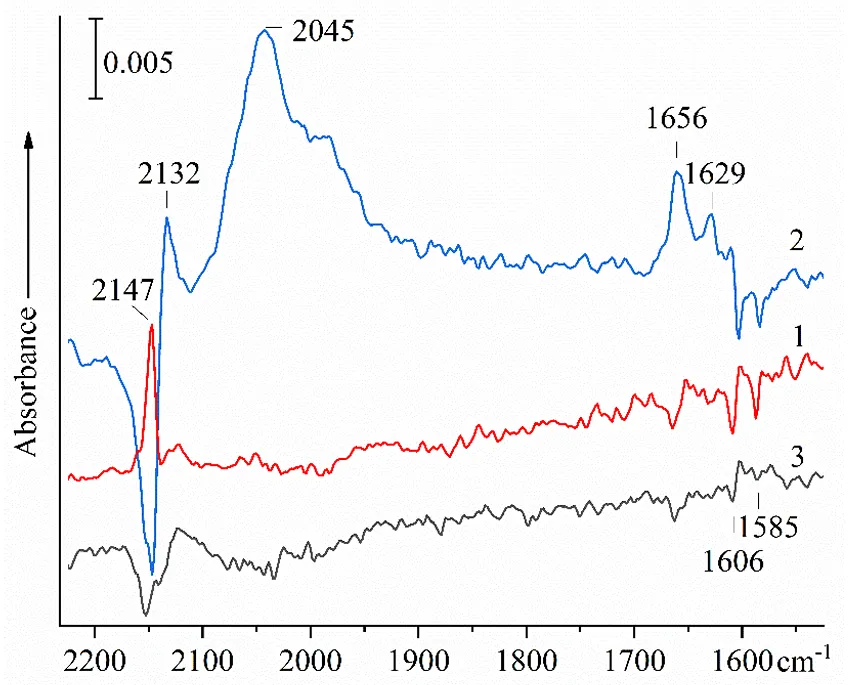
The results of a test experiment on the effect of resonant laser IR excitation on the spectrum of co-adsorbed HN
3 and DMP, described in [
25], are shown in
. Each curve in the figure represents the result of subtracting two absorption spectra recorded after a certain time interval from each other. It can be seen from the figure that standing the sample at −150 °C, when the vapor pressure of DMP and HN
3 is negligible, leads to insignificant changes in the spectrum. Serious changes, when the intensity of some bands decreases and others increase, occur during irradiation. The observed changes could be explained by the effect of a local temperature increase under the influence of laser radiation. Indeed, in both cases, the intensity of the 2050 cm
−1 bands and dimethylpyridinium ions increases. However, in the spectrum 1 in
, this is accompanied by intensity increase of 2147 cm
−1 band and decrease of that at 2136 cm
−1. After irradiation, on the contrary, the appearance of products occurs due to the consumption of compounds that absorb at 2147 cm
−1.
. Changes in the FTIR spectrum of aerosil SiO<sub>2</sub> pretreated at 600 °С with preliminary adsorbed at 20 DMP, addition of HN<sub>3</sub> at −80 °С and temperature stabilization for 20 min at −150 °С (<em>1</em>), irradiation at −150 °С for 30 min at 2140−2150 cm<sup>−1</sup> (<em>2</em>) and after keeping at the same temperature for 17 min (<em>3</em>). Each curve shows the difference between the spectra taken before and after the mentioned time interval [<a href="#B25" class="html-bibr">25</a>] (Reproduced by permission from <em>Optics and Spectroscopy</em>).
Test experiments on the effect of resonant laser IR radiation on the spectrum of co-adsorbed HN
3 and DMP have shown that the protonation process can be accelerated by resonant excitation of vibrations of HN
3 molecules, and the nature of the induced changes differs from those occurring spontaneously with increasing system temperature. In the latter case, the process of protonation of DMP occurs due to the consumption of HN
3 molecules responsible for the 2136 cm
−1 band, silanols interacting with oxygen associated with DMP, which is accompanied by deprotonation of acid molecules to form N
3− ions. The remaining molecules of adsorbed HN
3 contribute to the band 2147 cm
−1, the intensity of which increases. The effect of additional growth of the bands of dimethylpyridinium and N
3− ions caused by irradiation at a frequency of 2140–2150 cm
−1 with a decrease in the intensity of the bands of hydrogen-bound DMP at 1606 and 1585 cm
−1 is accompanied by a sharp decrease in the band 2147 cm
−1 and the increase in absorption at 2132 cm
−1, which is obviously the edge of the band 2136 cm
−1. Apparently, irradiation somehow promotes the interaction of HN
3 with the remaining hydrogen-bonded complexes of DMP with silanol groups, with further protonation of DMP and the formation of N
3− ions.
All laser irradiation experiments were carried out at the Resource Center of St. Petersburg State University Science Park “Centre for Optical and Laser Materials Research” (https://researchpark.spbu.ru/laser-rus). The authors are thankful to L. Khriachtchev (University of Helsinki, Finland) for the help in experiments with laser for 4300 cm−1 region and measurements at 8K.
Conceptualization, A.T. and O.P.; Methodology, A.T. and T.A.; Software, O.P.; Validation, A.T., O.P. and T.A.; Formal Analysis, A.T.; Investigation, O.P. and T.A.; Data Curation, O.P. and T.A.; Writing—A.T.; Visualization, O.P.; Supervision, A.T.; Project Administration, A.T.; Funding Acquisition, A.T.
Ethical review and approval are not applicable, since the study did not involve humans or animals.
Not applicable.
Data can be available on request.
This research was funded by RSCF grant number 24-23-00606, https://rscf.ru/project/24-23-00606/.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.