1. Introduction
The Han Chinese (HAN) are the largest ethnic group in the world, accounting for approximately 1/6 of the global population. However, a recent study noted that the vast majority (86%) of genomic studies were conducted on individuals of European descent [
1]. The field of genomics currently exhibits a serious imbalance in scientific research among different population groups. Of the 3202 genomes in the 1000 Genome Project, only 283 belong to the HAN, a proportion of 8.8% that is grossly disproportionate given the HAN’s share of the global population [
2,
3].
Knowledge about genetic structure can aid in inferring evolutionary history and guiding association studies [
4,
5,
6]. Although many studies have reported the overall picture of the genetic structure of the HAN population, more detailed population genetic structures are still worth improving, especially at the provincial, municipal, and minority ethnic group levels beyond the HAN population [
7,
8,
9,
10]. Moreover, in the past decade, there have been numerous population genetic studies utilizing genome-wide single nucleotide polymorphism (SNPs) and mtDNA analyses. Nevertheless, more comprehensive population studies are necessary to fully elucidate the intricate genetic structure of the patrilineal HAN.
A comprehensive analysis of mitochondrial DNA haplogroups on a large scale has revealed that the HAN population can be divided into three subgroups based on matrilineal ancestry: those inhabiting the Yellow River valley, the Yangtze River valley, and the Pearl River valley [
11]. In contrast, the genetic structure of the HAN obtained from genome-wide SNPs is much more intricate due to admixture. Previous studies conducted in China using genome-wide SNPs have revealed the potential existence of three to seven subgroups within the HAN population [
12,
13,
14,
15,
16]. These studies all accept that the HAN ethnic group can be categorized into three primary subgroups, with the Qinling-Huaihe River and the Nanling Mountains serving as the primary geographic boundaries, which is similar to the mtDNA study.
It is well-known that patrilineal HAN can be divided into two subgroups, North and South [
4,
17,
18]. Despite numerous studies on the fine-scale genetic structure at regional and provincial levels in China, a research gap still exists at a national level. Mainly because there is still a lack of a unified dataset to identify the intricate genetic structure within the subpopulations of North and South. Furthermore, population substructure can be complicated by several factors, including the number and types of genetic markers, geographic origins, and coverage of studied and reference populations [
19]. Therefore, conducting such research necessitates a unified dataset from a large population that takes into account both population coverage and high-density genetic markers.
Based on a 372 Y-chromosome haplogroup frequency dataset identified from 18,618 male individuals covering 33 provincial administrations in China, we present an overview of the Y chromosome haplogroup distributions of Han Chinese. Our study reveals that patrilineal HAN can be divided into five distinct geographic subgroups: Northern China, Eastern Coast, Upper and Middle Yangtze River, Lingnan, and Min-Tai. We also compared the frequency of mtDNA and Y haplogroups within the same sample size range, which helped us identify differences in the genetic structure of Han patrilineal and matrilineal populations and associate these differences with geographic factors. Specifically, we found that patrilineal HAN exhibited stronger geographic clustering compared to matrilineal populations. Our study provides a comprehensive analysis of the genetic substructure of patrilineal lineages of the HAN at a national level and highlights sex bias in the genetic structure with respect to patrilineal and matrilineal lineages. Additionally, we observed a decrease in haplogroup diversity (HD) from north to south in patrilineal HAN. Finally, our analysis of the NJ tree suggests a progressive relationship and indicates a southward migration route.
2. Results
2.1. An Overview of the Y Chromosome Haplogroup Distributions of the Han Chinese
Our dataset included a total of 372 haplogroups from 33 provincial-level administrative regions. The O haplogroup accounted for the highest proportion, with a prevalence of 78.06%, making it the most common Y-haplogroup among the patrilineal HAN (). Notably, the O2a2 haplogroup accounted for 36.2%, indicating that approximately 240 million males in China belong to this haplogroup (2021 Population Census). This was followed by the O2a1 haplogroup, accounting for 17.9%. Collectively, these two haplogroups accounted for 54.1% of the patrilineal HAN. In addition, several other haplogroups with a proportion greater than 1% were identified, including O1a1 (12.03%), O1b1 (9.39%), C (9.15%), N (6.17%), Q (2.78%), D (2.01%), O2b1 (1.20%), and R (1.02%) (). Additionally, other O haplogroups not included in the aforementioned haplogroups accounted for 1.34%. These haplogroups accounted for a cumulative prevalence of 99.19%, while the remaining nine haplogroups, A, E, F, G, H, I, J, L, and T, collectively accounted for only 0.81% and can be considered negligible. Therefore, our findings suggest that the O, C, N, Q, D, and R haplogroups are the predominant paternal haplogroups among the patrilineal HAN, which is consistent with the previous studies [
4,
20,
21,
22,
23,
24,
25,
26,
27,
28].
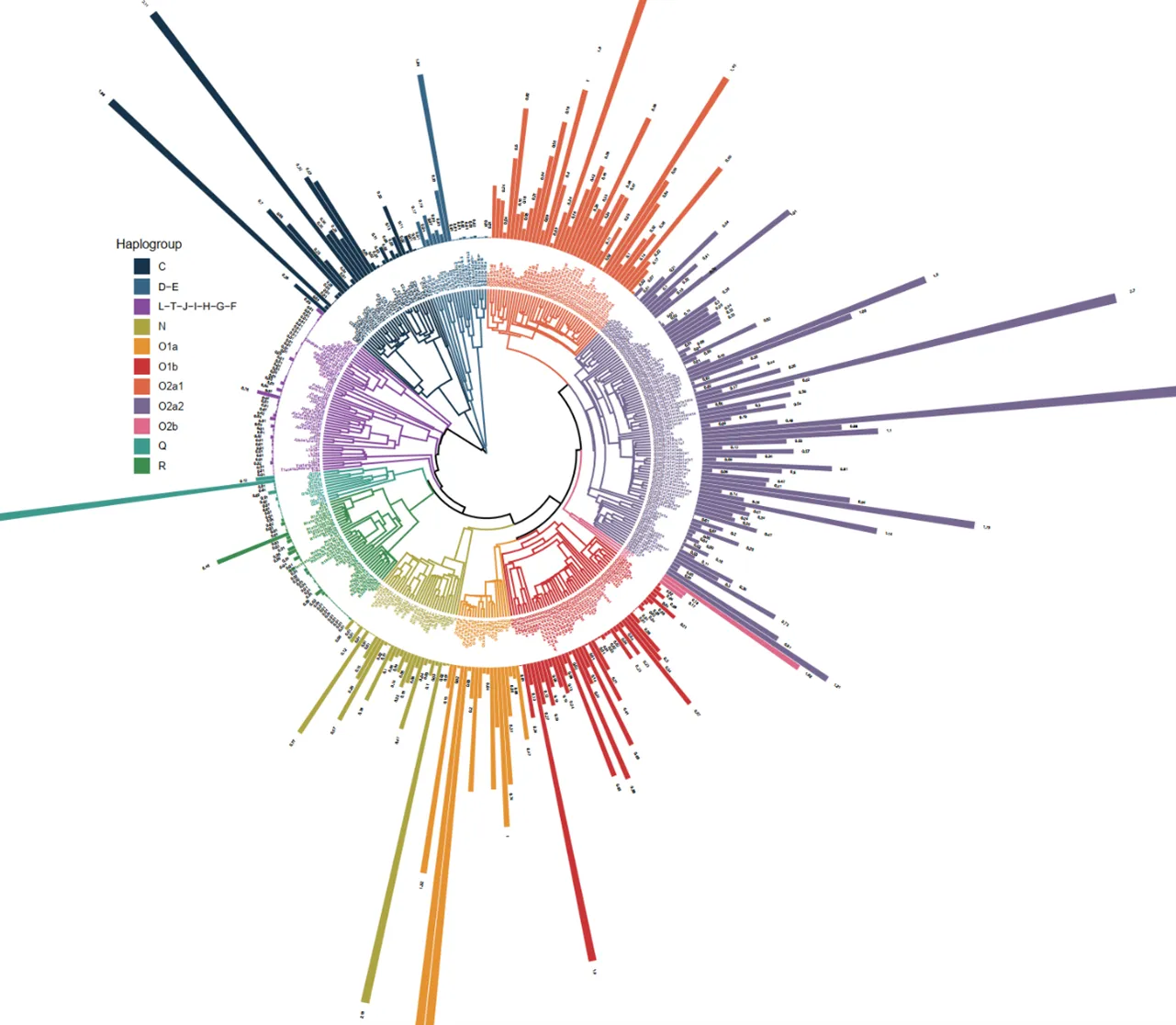
We constructed a phylogenetic tree of 372 haplogroups, which intuitively and clearly characterize the composition of the patrilineal HAN (). According to the frequency and phylogeny relationship, we divided the haplogroups into 11 major haplogroups, some of which showed “star-like” expansion, suggesting possible population expansion events, some of which have been previously reported, including the three super branches: O
α, O
β, and O
γ [
20].
2.2. The Location of the Patrilineal HAN in Worldwide Populations
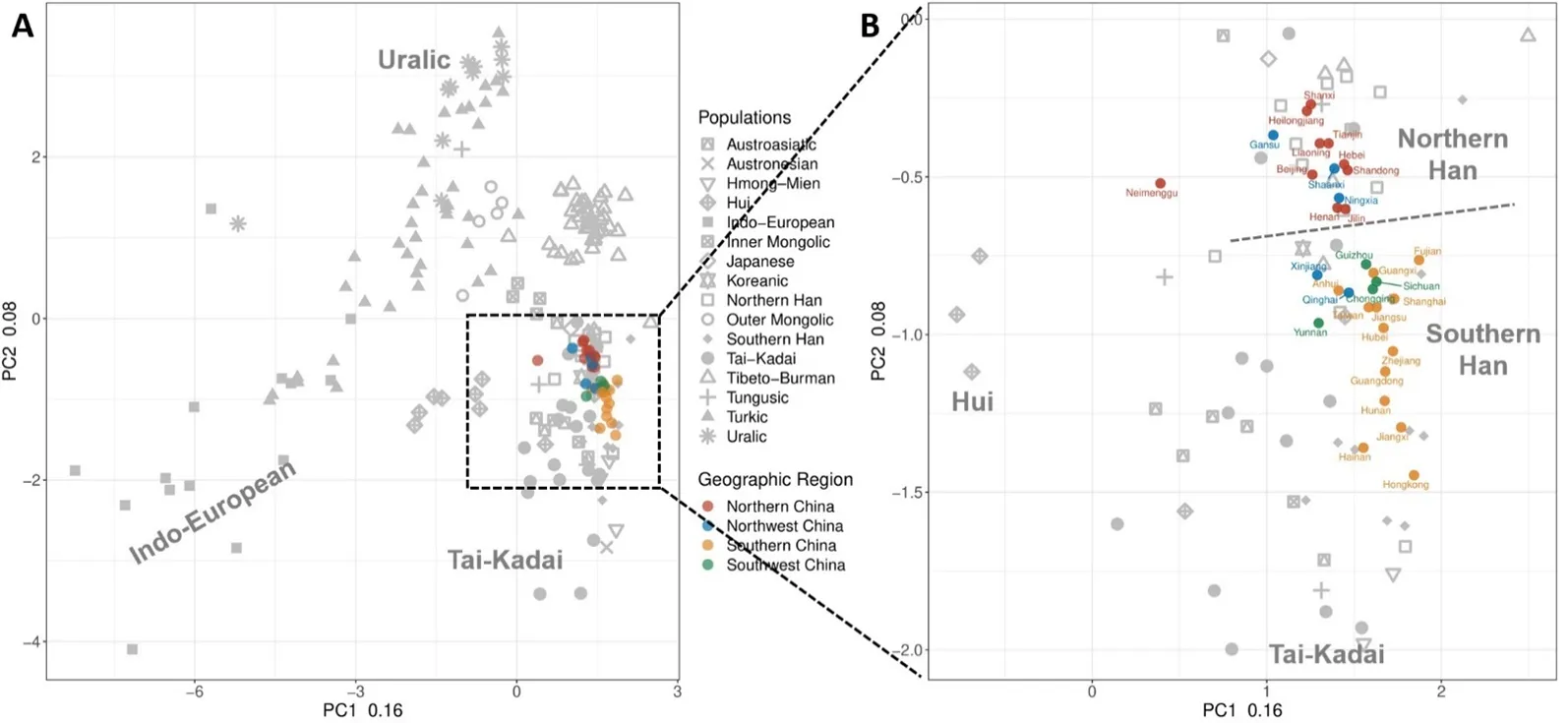
To determine the location of the patrilineal HAN in worldwide populations, we collected data on the frequency of 28 Y-haplogroups from 221 populations worldwide, including our dataset (Table S1). We performed principal component analysis (PCA), and the first two principal components (PCs) explained 16.1% and 7.9% of the total variance, respectively. PC1 was roughly correlated with the East-West direction, while PC2 was correlated with the North-South direction (A). Our analysis revealed a clear separation between different Eurasian populations, with the greatest divergence observed between Indo-European, Uralic, and Tai-Kadai groups. The genetic structure reflects their geographical position in Eurasia, with the patrilineal HAN is located in the core region of East Asia, surrounded by other East Asian populations such as the Japanese, Korean, and Tai-Kadai populations. This clustering result also suggests the genetic continuity of the patrilineal HAN. Within HAN, the provinces in northern China are separated from those in southern China (B). Our PCA results, as well as additional analyses using NJ-tree, 3D-PCA, and tSNE, all support the genetic continuity and North-South divergence of the Han Chinese (B,C; ; ).
. Phylogenetic tree was built from 372 haplogroups identified in the patrilineal HAN. Each tips represents a haplogroup, and colors are used to distinguish 11 macro-haplogroups. The coalescent time of the node is referenced to Yfull database, and the root node represents the coalescence time of about 69,000 years ago. The length of the outer band and the black number represent the percentage of the haplogroup in all samples.
. PCA plots of 221 global populations based on 28 Y-haplogroups. (A) PCA for 221 global populations. Public data populations have been shown in grey dots in the background, and the HAN data generated from this study color-labelled by geographic region. (B) Partially magnified PCA emphasizes the HAN population. Colors indicate different geographical areas. red: Northern HAN; blue: Northwest HAN; yellow: Southern HAN; green: Southwest HAN. The genetic division of the Southern and Northern populations is clearly visible.
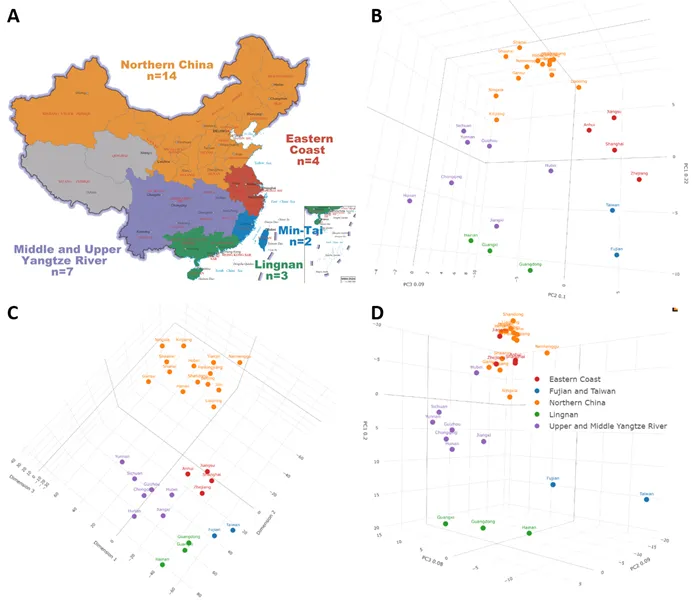
. Fine-scale genetic substructure of the HAN. Color is used to characterize different subgroups. For three-dimensional clustering details, see Supplementary files S1-S4. (A) Genetic structure of the patrilineal HAN population illustrated with a map, genetic structure results from PCA. Both PCA and tSNE showed that the patrilineal HAN could be divided into five subgroups: Northern China, Eastern Coast, Upper and Middle Yangtze River, Lingnan, and Min-Tai (Map Inspection Number: GS (2019)1682). (B) The PCA clustering based on 372 Y-haplogroups in HAN. (C) The clustering results of tSNE, with a slightly difference of PCA. (D) PCA based on 854 mtDNA haplogroups in HAN which reflects matrilineal genetic structure.
To explore the fine-scale genetic structure of the patrilineal HAN, PCA and tSNE were used. We excluded Macau, Hong Kong, and Qinghai provinces due to small sample sizes (<30). Our results revealed that the patrilineal HAN can be divided into five distinct subgroups: Northern China, Eastern Coast, Upper and Middle Yangtze River, Lingnan, and Min-Tai (Fujian and Taiwan) (A–C; Supplementary files S1-S2). The Northern China subgroup included all 14 northern provinces, while the Eastern Coast subgroup was comprised of four provinces downstream of the Yangtze River and on the Eastern Coast area (Jiangsu, Zhejiang, Shanghai, and Anhui). The Upper and Middle Yangtze River subgroup was comprised of seven provinces along the Yangtze River, excluding the aforementioned downstream provinces. The Min-Tai subgroup included Fujian and Taiwan provinces, while the Lingnan subgroup included Guangdong, Guangxi, and Hainan provinces (A).
PCA analysis revealed that PC1, PC2, and PC3 accounted for 22.2%, 10.2%, and 9.2% of the total variance, respectively. The five subgroups in the PCA corresponded well with their actual geographic locations, with PC1 reflecting the North-South cline and PC2 roughly separating East and West China. The tSNE clustering was consistent with the PCA, as well as the consistency of each subgroup’s composition (C; Supplementary files S1 and S2). The lack of a linear relationship between dimensions is an essential feature of the tSNE algorithm. However, a strong correlation existed between the groups in the tSNE dimensions and real geographic relationship, particularly in the first two dimensions, closely matched the geography of Chinese provinces. These indicating a complex, nonlinear relationship between geographical factors and Y-chromosome haplogroup frequencies, which could not be explained by latitude and longitude alone.
To avoid bias in the comparison of Y-chromosome and mtDNA results, we downloaded an mtDNA dataset with almost the same sample size as ours (21668) from a published literature [
11]. Identical PCA and tSNE were used to identify the genetic structure. In PCA, PC1, PC2, and PC3 explained 20.2%, 9.5%, and 7.9% of the variance, respectively. PC1 showed a clear North-South geographic orientation, consistent with the Y chromosome results (D; Supplementary files S3 and S4), while PC2 and PC3 did not (Figure S2). Our PCA results indicated that the matrilineal HAN could be divided into three to five subgroups, but the subgroup boundaries were not as clear-cut as those in the patrilineal HAN. Provinces within the subgroups were geographically far apart, with Ningxia and Taiwan provinces distant from other subgroups and almost appearing as separate subgroups. Different from PCA, tSNE provided more distinct clustering findings than PCA, with distinct boundaries among the three subgroups and no outliers (D; Supplementary files S3 and S4). tSNE and PCA results suggested that the matrilineal HAN may have had more population mixing events than patrilineal HAN, resulting in a blur of subgroup boundaries. The haplogroup diversity analysis yielded similar results (, Table S11).
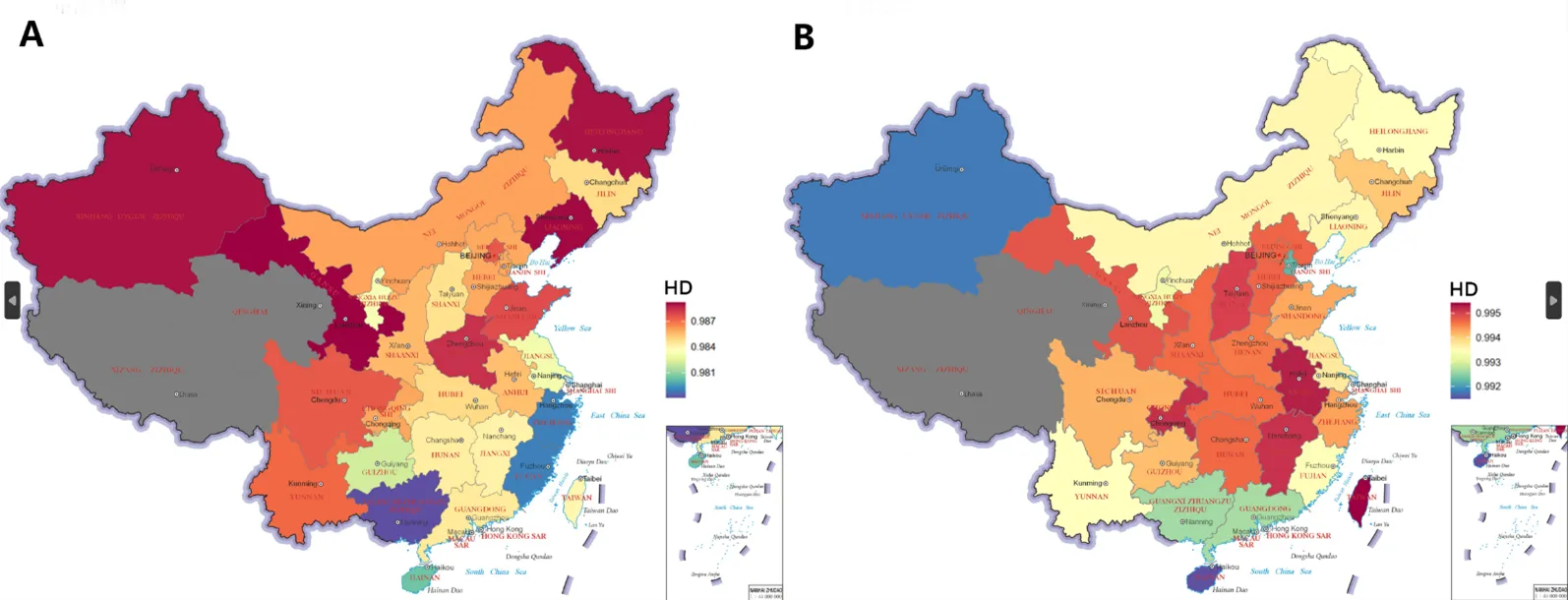
. Haplogroup diversity results presented in map. Higher HD values indicated in red. Lower HD values indicated in blue. Provinces in grey indicate no sample coverage. (Map Inspection Number: GS (2019)1682). (A) Haplogroup diversity map for patrilineal HAN. (B) Haplogroup diversity map for matrilineal HAN.
To confirm the fine-scale genetic structure, we calculated the inter-group variation using AMOVA with various classification schemes (Table S2). We classified the HAN population into 1–8 subgroups (Table S2). The inter-group variation reflects the distance between different subgroups. These AMOVA results confirmed that the most significant inter-group variation was 1.60%, when using a five subgroup scheme for patrilineal HAN (). According to AMOVA, inter-group variation was also significant (1.41%) when the population was classified into three subgroups: Northern China, Yangtze River, and Zhujiang River. In comparison, the inter-group variation for HAN in Southern and Northern China was 1.17%, and there was considerable variation within group (). Notably, dividing the Northern China group into two subgroups resulted in an inter-group variation of 1.57%, approaching the largest inter-group variation (1.60%). This grouping pattern was consistent with the clustering results of PCA and tSNE, adding further support to the five subgroups within the patrilineal HAN.
Similar to the patrilineal population, AMOVA was utilized to examine the inter-group variations in matrilineal HAN. When we divided matrilineal HAN into four subgroups based on PCA, we observed the greatest inter-group difference, at 0.995%. Inter-group variation stood at 0.981% using three subgroups according to tSNE (Table S3). These two values were similar and both markedly greater than other classification schemes, such as the five subgroup scheme based on the Y chromosome, which only demonstrated 0.613% inter-group variance (Table S3). This suggested that patrilineal and matrilineal HAN have different genetic substructures. Moreover, different subgroup patterns demonstrated by tSNE and PCA, as supported by AMOVA, reveal the complex genetic structure of matrilineal HAN. It appears that the genetic boundaries of matrilineal HAN are not as well-defined as those in patrilineal HAN.
Comparison with Y haplogroup, AMOVA based on mtDNA demonstrated larger intra-population and smaller inter-group variation on average. The intra-population variation reflects the similarity between individuals within the population. When the HAN was considered as one group, intra-population variation of mtDNA stood at 99.06% and at 98.33% of Y chromosome (; Table S3), indicating that matrilineal HAN was more closely connected at individual level by comparison with patrilineal HAN. This also explained the blurred boundary among matrilineal subgroups discussed above. These findings were further supported by the subsequent HD and NJ tree results. The observation of mixed matrilineal HAN and more distinct patrilineal HAN suggests a sex bias in Han Chinese populations.
. AMOVA based on Y haplogroups under various classification schemes. For further details, see supplementary Table S2 and S3.
PCA, tSNE, and AMOVA revealed a relationship between the genetic structure and geography. For quantitative characterization, we calculated the correlation between PCs and geography (Figure S4; Figure S1). In addition, we utilized a Mantel test to quantify the correlation between geographical distance and genetic distance.
PC1 and latitude had the greatest Pearson correlation coefficient (PCCs) of 0.86 (
p-value = 0.001) in the Y chromosome PCA (Figure S2C), while PC2 and longitude had a relatively lower correlation of 0.64 (
p-value = 0.001) (Figure S2B). In the mtDNA PCA, PC1 had the highest PCCs of −0.85 (
p-value 0.001), while PC2 and longitude had only 0.03 PCCs and was not statistically significant (
p-value = 0.857) (Figure S3). Regardless of patrilineal or matrilineal lineage, the above results indicate that the latitude, or North-South direction, was the primary factor determining the genetic structure of the Chinese population. Longitudinal East-West direction significantly influenced the genetic structure of the patrilineal HAN but had little effect on the matrilineal HAN. This trend has been overlooked in prior investigations with limited sample sizes. Similarly, we observed that the PCs correlation on average was slightly higher in the patrilineal HAN as opposed to the matrilineal HAN, suggesting the possibility of stronger geographic correlation among patrilineal HAN.
We also conducted a Mantel test to examine the relationship between the geographic and genetic distance (Table S4). Here, the correlation between geographic distance (Vincenty great circle distance) and genetic distance (pairwise Fst, Euclidean distance in PC space) was greater for patrilineal HAN, indicating more intense geographical concentration. For example, the correlation of Fst and Vincent distance for the Y chromosome was 0.4597 and 0.3610 for mtDNA. Moreover, we observed different geographical patterns between the patrilineal and matrilineal HAN in southern and northern provinces, respectively. Specifically, for the Y chromosome, the correlation was stronger in the northern provinces (0.4351) than in the southern provinces (0.3322) (Table S4), indicating the geographical environment in the northern region has shaped a more homogeneous paternal lineage. In contrast, for mtDNA results, the correlation was a little weaker in the northern provinces (0.2302) than in the southern provinces (0.2649). These results suggested that the geography of northern China has a greater impact on patrilineal HAN, while the geography of southern China exerted a greater impact on matrilineal HAN. These results reflect a geographical sex bias in the Han Chinese.
2.6. Higher Haplogroup Diversity of Northern HAN Indicates North to South Migration
We calculated haplogroup diversity (HD) values for the patrilineal and matrilineal HAN for each province. We found that, on average, the matrilineal HAN exhibit higher HD values than patrilineal HAN. Specifically, the average HD value for patrilineal HAN was 0.985, while the value for matrilineal HAN was 0.994 (A,B).
In terms of geographic distribution, provinces with higher HD value of patrilineal HAN were mainly located in frontier regions such as northern China, southwestern China, and Xinjiang (A). In contrast, the matrilineal HAN showed different pattern, showing higher values in central China (Henan, Hubei, Anhui, and Jiangxi) (B). Interestingly, Henan, representing central China, exhibited higher haplogroup diversity for both Y chromosome and mtDNA. In central China, the higher haplogroup diversity of mtDNA probably due to the convergence of populations from surrounding regions. In border regions of China, the high diversity of Y chromosome may be the result of admixture with minority and Central Plains HAN, although this process may not have involved matrilineal populations. The lower haplogroup diversity of southern HAN may be due to genetic bottlenecks experienced during historical migrations from north to south.
2.7. NJ Tree Indicates a Possible Southward Migration Route of HAN
We calculated the Euclidean distance matrix and the pairwise F
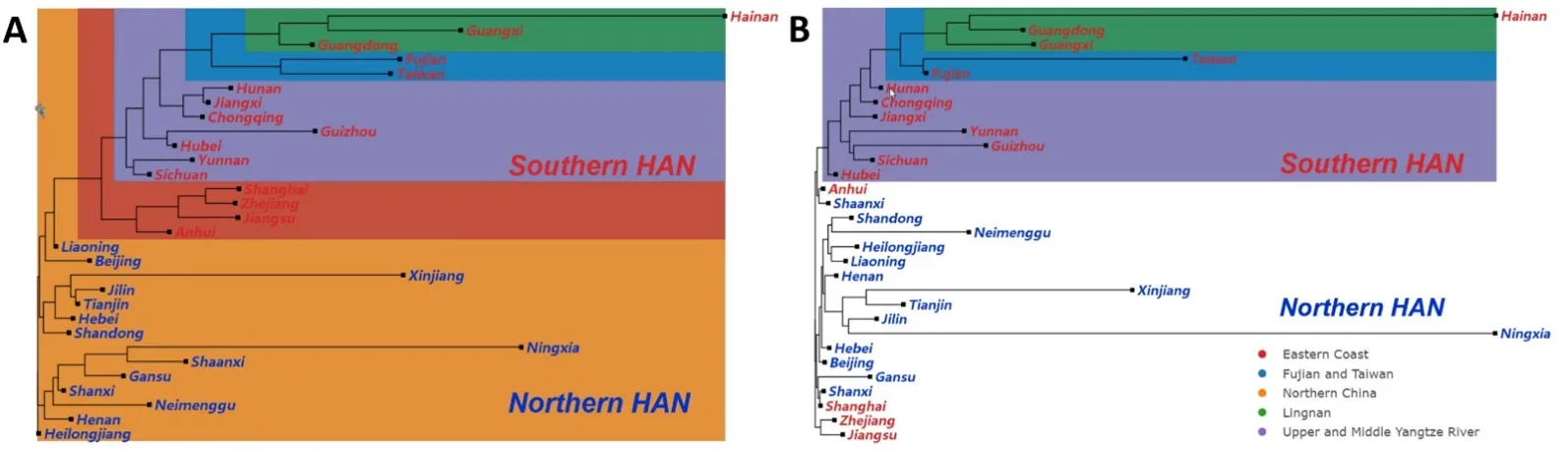
st distance matrix (Table S6; Table S11) between each province. Based on these two matrices, we constructed NJ trees for the patrilineal and matrilineal HAN, respectively (; Figures S5 and S6). Our results revealed a clear separation between the southern and northern patrilineal HAN, with both distance-based NJ trees exhibitisng two distinct clades (; Figures S5 and S6). However, within the same clade, the matrilineal HAN showed some intermingling between southern and northern provinces. Meanwhile, our analysis demonstrated that the northern patrilineal HAN were more closely clustered than their southern counterparts, consistent with the PCA findings ().
Interestingly, previous studies have revealed a male-dominated north-to-south migration of Chinese populations throughout history [
18]. Our haplogroup diversity results supported this conclusion (). The NJ tree also exhibited a stepped structure with four endpoints for north-to-south migration. Starting with the northern population on the NJ tree, the Eastern Coast subgroup was the first to diverge, followed by the seven provinces of the Upper and Middle Yangtze River subgroup, and finally Fujian-Taiwan and the three Lingnan provinces. This indicated that northern inhabitants may have migrate in Lingnan and Fujian-Taiwan via the middle Yangtze River, as opposed to the Eastern Coast. The PCA confirmed this result. According to PCA, the Lingnan and the Upper and Middle Yangtze River subgroups exhibited closest proximity as well (B).
. Unrooted Neighbor-Joining (NJ) trees of HAN based on pairwise Fst values. (A) NJ-tree based on the Y-haplogroup frequency for each province in China. (B) NJ-tree based on the mtDNA haplogroup frequency of each province in China. The colored box shows the subgroups referred to in the text. Orange indicates the Northern China subgroups, red indicates the Eastern Coast, purple indicates the Upper and Middle Yangtze River, blue indicates Lingnan, and green indicates Min-Tai.
3. Discussion
Although the genetic structure of the Han population has been extensively studied, balancing sample size and marker density remains a challenge. In this work, a large sample size covering almost all of China was used to discuss the genetic structure of the patrilineal HAN with high density markers of Y chromosome genotyping. It should be noted that sampling bias affects the accuracy of the genetic structure, which leads to two problems. The first is that the estimation of haplogroup frequency in provinces with fewer samples, such as Ningxia and Xinjiang, would be impacted, leading to a deviation from the main cluster. The second limitation is that despite having a sample size of nearly 20,000 individuals, it may not be fully representative of the patrilineal Han population in China, given the country’s vast population size.
Our study revealed a clearer genetic structure among patrilineal HAN compared to matrilineal HAN. This structure was characterized by identifiable group boundaries and lower haplogroup diversity, suggesting a “sex bias” in genetic inheritance. Additionally, patrilineal HAN exhibited fewer individual differences within each group and a better spatial aggregation, which implies fewer genetic founders and the migration bottleneck. Regrettably, genomic data on individual-level of mtDNA is currently unavailable, which limits the precision of comparing the geographical distribution patterns between patrilineal and matrilineal HAN. Conducting a quantitative analysis based on individual genomic variations may yield more accurate explanations or even challenge existing conclusions.
Our objective is to establish a macroscopic migration route for the patrilineal HAN, rather than focusing solely on the a single haplogroup lineage. While individual-level or haplogroup-level phylogenetic trees may provide more detailed insights than the average at the provincial level, clearly explained the history of each haplogroup is a complex issue. For instance, the O haplogroup has three major lineages, along with minor ones, that coalescent around 6000–6500 years before present and are dispersed throughout China via intricate origin and migration paths [
20]. Additionally, populations in different haplogroups may exhibit distinct or even opposing migration directions, which could complicate the overall migration route of the HAN. While this study may not provide a detailed analysis of each haplogroup, provides valuable references, and we intend to examine the origin and migration paths of each haplogroup in future studies, which will become a more complete use of the dataset.
Lastly, the results of the NJ tree demonstrated a progressive relationship, suggesting a southward migration route. We speculate that the HAN arrived in the Yangtze River valley from the north before further spreading along the middle reaches of the Yangtze River to Fujian, Taiwan, and Lingnan, rather than via the East China Coast. This result is also more consistent with the geography of China, as the mountainous terrain of Fujian and Zhejiang may have impeded the population’s spread. In summary, our study provides valuable insights into the basic genetic landscape of the patrilineal HAN and sheds light on the historical migration patterns of Chinese populations.
4. Data and Methods
4.1. Data
Data1: Our database contains haplogroup frequency for 18618 individuals from 33 provincial-level administrations in China. The data contained a total of 372 Y-chromosome haplogroups and native provinces. The haplogroup definition of these makers was strictly adopted according to the International Society of Genetic Genealogy (ISOGG-2019.10.1).
Data2: We collected frequency data for 28 Y-chromosome haplogroups from 189 populations worldwide from published literatures [
9,
10,
29,
30,
31,
32,
33,
34,
35,
36,
37,
38,
39,
40,
41,
42,
43,
44,
45,
46,
47,
48,
49,
50,
51,
52]. We coalesced haplogroups from our Chinese population internal database into 28 macro-haplogroups for analysis, for a total of 221 groups included. This dataset is available in Table S1.
Data3: We downloaded mtDNA haplogroup frequency data for HAN from published literature, which included 21668 individuals from 33 provincial-level administrations in China [
11]. A total of 854 mtDNA haplogroups were identified. This dataset is available in Table S5.
4.2. Methods
Unless specifically emphasized, the statistical analyses in this study were performed in
R-4.0.5 environment.
4.2.1. Dimensionality Reduction
PCA and tSNE: We used two data dimensionality reduction methods to cluster the frequency data of mtDNA and Y-chromosome haplogroups separately so as to uncover the implied subgroup. A linear method of dimensionality reduction (PCA) and a non-linear method of dimensionality reduction (tSNE) were used as comparison. The PCA in this study was conducted using the
PCA function in the
FactoMineR package in
R-4.0.5. The t-distributed Stochastic Neighbor Embedding (t-SNE) was conducted using
Rtsne 0.16 package.
4.2.2. Distance Calculation
F
st: Genetic distances (F-statistics, Fst) based on the haplogroup frequencies were calculated by
Arlequin 3.5.2.2. We calculated pair-F
st matrices for the patrilineal and matrilineal HAN in each province separately (Table S6).
Vincenty distance: The geographic distances are characterized using the Vincenty great circle distance (Tables S7 and S8), which is based on the assumption of an oblate spheroid earth, improving accuracy compared to great-circle distance, which assumes a spherical Earth. The latitude and longitude of each province are represented by provincial capital (Table S9). The Vincenty distance was conducted using
geosphere 1.5-14 packages.
Euclidean distance in PCA space: We calculated the Euclidean distances of the points represented by each province in the 3D-PCA space using the
dist function in R (Table S10).
Euclidean distance based on haplogroup frequency: We calculated the paired Euclidean distances by each province based on the haplogroup frequency data using the
dist function in R (Table S11).
4.2.3. Population Classification and AMOVA
We classified the Han Chinese in each province, based on previous studies and the clustering results in this study. We used 19 classifications to place the HAN population into 1–8 different subgroups (Table S2). Analysis of molecular variance (AMOVA) based on haplogroup frequencies was used to test the precision of clustering results (; Table S3). Generally, a larger inter-group variation translated to a better clustering result. AMOVA was conducted using
ade4 1.7-19 packages in R.
4.2.4. Haplogroup Diversity
Haplogroup diversity (
HD) is a measurement of the uniqueness of a particular haplogroup in a given population. HD was gauged by the formula:
where,
n denoted the sample size, and
xi represented the frequency of a haplogroup within a province [
53]. We calculated the Y chromosome and mtDNA haplogroup diversity of HAN in each province separately, as detailed in Table S12.
4.2.5. Geographical Aggregation and Mantel Test
We assessed the correlation between principal components (PCs) and geography in the patrilineal and matrilineal HAN, respectively. Specifically, the
cor function was used to calculate the Pearson correlation coefficient (PCCs) between PCs and latitude and longitude (Figures S1 and S2).
Subsequently, we performed the Mantel test in order to quantitatively represent the relationship between genetic and geographical distances and to assess sex differences (Table S4). The Mantel test is a correlation test that determines the correlation between two matrices. Here the function was performed to check the correlation between geographic distance matrices (Vincenty distance) and genetic distance matrices (F
st/Euclidean distance in 3D-PCA space). Our Mantel test was conducted using
vegan 2.6-2 packages.
4.2.6. Unrooted Neighbor-joining Tree
We conducted the unrooted neighbor-joining (NJ) trees based on population pairwise Fst value and Euclidean distance of haplogroup frequency (; Figures S3 and S4). The NJ tree was conducted using the
ape 5.6-2 package and Euclidean distance was calculated by the
dist function in R-4.0.5.
Supplementary Materials
Supporting information can be found at: https://www.sciepublish.com/index/journals/article/natanthropol/34/id/53.
Acknowledgments
The investigators thank all the donors for providing biological samples. We thank Chengdu 23MoFang, Inc. for genotyping and provide data.
Author Contributions
Yichen Tao: Conceptualization, Methodology, Software, Formal analysis, Visualization, Writing-Original draft, Writing-Review & Editing. Zhou Juanjuan: Writing - Original draft, Validation, Investigation, Data curation. Edward Allen: Writing-Review & Editing. Letong Liang: Writing-Review & Editing, Validation, Formal analysis. Yetao Zou: Data curation. Zishuai Huang: Data curation. Hui Li: Data curation, Writing-Review & Editing, Supervision, Project administration, Funding acquisition.
Ethics Statement
The conception and implementation of this study were originally approved by the Ethics Committee of Fudan University (code: 14012) and retrieval of genetic data was approved in 2023 (FE23187I).
Informed Consent Statement
Participants signed online consent forms before participating in the study and data producer proceeded in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans according to the declarations of the data producer and project supervisors. The study was also conducted in accordance with the Principles of Human and Ethical Research of the Ministry of Science and Technology of the People’s Republic of China (Interim Measures for the Management of Human Genetic Resources, June 10, 1998).
Funding
This work was supported by the National Key R&D Program of China (2020YFE0201600), B&R Joint Laboratory of Eurasian Anthropology (18490750300), and the National Natural Science Foundation of China (91731303 and 31671297 to Hui Li). The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Declaration of Competing Interest
The authors declare no conflict of interest.
References
-
1.
Fatumo S, Chikowore T, Choudhury A, Ayub M, Martin AR, Kuchenbaecker K. A roadmap to increase diversity in genomic studies.
Nat. Med. 2022,
28, 243–250.
[Google Scholar]
-
2.
Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation.
Nature 2015,
526, 68–74.
[Google Scholar]
-
3.
Fairley S, Lowy-Gallego E, Perry E, Flicek P. The International Genome Sample Resource (IGSR) collection of open human genomic variation resources.
Nucleic Acids Res. 2020,
48, D941–D947.
[Google Scholar]
-
4.
Xue FH, Wang Y, Xu SH, Zhang F, Wen B, Wu XS, et al. A spatial analysis of genetic structure of human populations in China reveals distinct difference between maternal and paternal lineages.
Eur. J. Hum. Genet. 2008,
16, 705–717.
[Google Scholar]
-
5.
Xue Y, Zerjal T, Bao W, Zhu S, Shu Q, Xu J, et al. Male demography in East Asia: a north-south contrast in human population expansion times.
Genetics 2006,
172, 2431–2439.
[Google Scholar]
-
6.
Rosenberg NA, Pritchard JK, Weber JL, Cann HM, Kidd KK, Zhivotovsky LA, et al. Genetic Structure of Human Populations.
Science 2002,
298, 2381–2385.
[Google Scholar]
-
7.
Ma X, Yang W, Gao Y, Pan Y, Lu Y, Chen H, et al. Genetic Origins and Sex-Biased Admixture of the Huis.
Mol. Biol. Evol. 2021,
38, 3804–3819.
[Google Scholar]
-
8.
Pan Y, Wen J, Ning Z, Yuan Y, Liu X, Yang Y, et al. Comparative Genomic and Transcriptomic Analyses Reveal the Impacts of Genetic Admixture in Kazaks, Uyghurs, and Huis.
Mol. Biol. Evol. 2023,
40, msad054.
[Google Scholar]
-
9.
Yin C, Su K, He Z, Zhai D, Guo K, Chen X, et al. Genetic Reconstruction and Forensic Analysis of Chinese Shandong and Yunnan Han Populations by Co-Analyzing Y Chromosomal STRs and SNPs.
Genes 2020,
11, 743.
[Google Scholar]
-
10.
Yao H, Wang M, Zou X, Li Y, Yang X, Li A, et al. New insights into the fine-scale history of western-eastern admixture of the northwestern Chinese population in the Hexi Corridor via genome-wide genetic legacy.
Mol. Genet. Genom. 2021,
296, 631–651.
[Google Scholar]
-
11.
Li YC, Ye WJ, Jiang CG, Zeng Z, Tian JY, Yang LQ, et al. River Valleys Shaped the Maternal Genetic Landscape of Han Chinese.
Mol. Biol. Evol. 2019,
36, 1643–1652.
[Google Scholar]
-
12.
Xu S, Yin X, Li S, Jin W, Lou H, Yang L, et al. Genomic dissection of population substructure of Han Chinese and its implication in association studies.
Am. J. Hum. Genet. 2009,
85, 762–774.
[Google Scholar]
-
13.
Chiang CWK, Mangul S, Robles C, Sankararaman S. A Comprehensive Map of Genetic Variation in the World’s Largest Ethnic Group-Han Chinese.
Mol. Biol. Evol. 2018,
35, 2736–2750.
[Google Scholar]
-
14.
Gao Y, Zhang C, Yuan L, Ling Y, Wang X, Liu C, et al. PGG.Han: the Han Chinese genome database and analysis platform.
Nucleic Acids Res. 2020,
48, D971–D976.
[Google Scholar]
-
15.
Li L, Huang P, Sun X, Wang S, Xu M, Liu S, et al. The ChinaMAP reference panel for the accurate genotype imputation in Chinese populations.
Cell Res. 2021,
31, 1308–1310.
[Google Scholar]
-
16.
Cong PK, Bai WY, Li JC, Yang MY, Khederzadeh S, Gai SR, et al. Genomic analyses of 10,376 individuals in the Westlake BioBank for Chinese (WBBC) pilot project.
Nat. Commun. 2022,
13, 2939.
[Google Scholar]
-
17.
Chu JY, Huang W, Kuang SQ, Wang JM, Xu JJ, Chu ZT, et al. Genetic relationship of populations in China.
Proc. Natl. Acad. Sci. USA 1998,
95, 11763–11768.
[Google Scholar]
-
18.
Wen B, Li H, Lu D, Song X, Zhang F, He Y, et al. Genetic evidence supports demic diffusion of Han culture.
Nature 2004,
431, 302–305.
[Google Scholar]
-
19.
Sul JH, Martin LS, Eskin E. Population structure in genetic studies: Confounding factors and mixed models.
PLoS Genet. 2018,
14, e1007309.
[Google Scholar]
-
20.
Yan S, Wang C-C, Zheng H-X, Wang W, Qin Z-D, Wei L-H, et al. Y Chromosomes of 40% Chinese Descend from Three Neolithic Super-Grandfathers.
PLoS ONE 2014,
9, e105691.
[Google Scholar]
-
21.
Sun N, Ma PC, Yan S, Wen SQ, Sun C, Du PX, et al. Phylogeography of Y-chromosome haplogroup Q1a1a-M120, a paternal lineage connecting populations in Siberia and East Asia.
Ann. Hum. Biol. 2019,
46, 261–266.
[Google Scholar]
-
22.
Wang L-X, Lu Y, Zhang C, Wei L-H, Yan S, Huang Y-Z, et al. Reconstruction of Y-chromosome phylogeny reveals two neolithic expansions of Tibeto-Burman populations.
Mol. Genet. Genom. 2018,
293, 1293–1300.
[Google Scholar]
-
23.
Lu Q, Cheng HZ, Li L, Yao HB, Ru K, Wen SQ, et al. Paternal heritage of the Han Chinese in Henan province (Central China): high diversity and evidence of in situ Neolithic expansions.
Ann. Hum. Biol. 2020,
47, 294–299.
[Google Scholar]
-
24.
Wang C-C, Yan S, Qin Z-D, Lu Y, Ding Q-L, Wei L-H, et al. Late Neolithic expansion of ancient Chinese revealed by Y chromosome haplogroup O3a1c-002611.
J. Syst. Evol. 2013,
51, 280–286.
[Google Scholar]
-
25.
Zhong H, Shi H, Qi XB, Xiao CJ, Jin L, Ma RZ, et al. Global distribution of Y-chromosome haplogroup C reveals the prehistoric migration routes of African exodus and early settlement in East Asia.
J. Hum. Genet. 2010,
55, 428–435.
[Google Scholar]
-
26.
Shi H, Zhong H, Peng Y, Dong YL, Qi XB, Zhang F, et al. Y chromosome evidence of earliest modern human settlement in East Asia and multiple origins of Tibetan and Japanese populations.
BMC Biol. 2008,
6, 45.
[Google Scholar]
-
27.
Xue Y, Zerjal T, Bao W, Zhu S, Shu Q, Xu J, et al. Male demography in East Asia: a north-south contrast in human population expansion times.
Genetics 2006,
172, 2431–2439.
[Google Scholar]
-
28.
Cai X, Qin Z, Wen B, Xu S, Wang Y, et al. Human migration through bottlenecks from Southeast Asia into East Asia during Last Glacial Maximum revealed by Y chromosomes.
PLoS ONE 2011,
6, e24282.
[Google Scholar]
-
29.
Al-Zahery N, Pala M, Battaglia V, Grugni V, Hamod MA, Hooshiar Kashani B, et al. In search of the genetic footprints of Sumerians: a survey of Y-chromosome and mtDNA variation in the Marsh Arabs of Iraq.
BMC Evol. Biol. 2011,
11, 288.
[Google Scholar]
-
30.
Di Cristofaro J, Pennarun E, Mazières S, Myres NM, Lin AA, Temori SA, et al. Afghan Hindu Kush: where Eurasian sub-continent gene flows converge.
PLoS ONE 2013,
8, e76748.
[Google Scholar]
-
31.
Dudás E, Vágó-Zalán A, Vándor A, Saypasheva A, Pomozi P, Pamjav H. Genetic history of Bashkirian Mari and Southern Mansi ethnic groups in the Ural region.
Mol. Genet. Genom. 2019,
294, 919–930.
[Google Scholar]
-
32.
Dulik MC, Osipova LP, Schurr TG. Y-chromosome variation in Altaian Kazakhs reveals a common paternal gene pool for Kazakhs and the influence of Mongolian expansions.
PLoS ONE 2011,
6, e17548.
[Google Scholar]
-
33.
Dulik MC, Zhadanov SI, Osipova LP, Askapuli A, Gau L, Gokcumen O, et al. Mitochondrial DNA and Y chromosome variation provides evidence for a recent common ancestry between Native Americans and Indigenous Altaians.
Am. J. Hum. Genet. 2012,
90, 229–246.
[Google Scholar]
-
34.
Karafet TM, Osipova LP, Savina OV, Hallmark B, Hammer MF. Siberian genetic diversity reveals complex origins of the Samoyedic-speaking populations.
Am. J. Hum. Biol. 2018,
30, e23194.
[Google Scholar]
-
35.
Khar’kov VN, Khamina KV, Medvedeva OF, Shtygasheva OV, Stepanov VA. Genetic diversity of Khakassian gene pool: subethnic differensiation and the structure of Y-chromosome haplogroups.
Mol. Biol. (Mosk.) 2011,
45, 446–458.
[Google Scholar]
-
36.
Khar’kov VN, Medvedeva OF, Luzina FA, Kolbasko AV, Gafarov NI, Puzyrev VP, et al. Comparative characteristics of the gene pool of Teleuts inferred from Y-chromosomal marker data.
Genetika 2009,
45, 1132–1142.
[Google Scholar]
-
37.
Kutanan W, Shoocongdej R, Srikummool M, Hübner A, Suttipai T, Srithawong S, et al. Cultural variation impacts paternal and maternal genetic lineages of the Hmong-Mien and Sino-Tibetan groups from Thailand.
Eur. J. Hum. Genet. 2020,
28, 1563–1579.
[Google Scholar]
-
38.
Lang M, Liu H, Song F, Qiao X, Ye Y, Ren H, et al. Forensic characteristics and genetic analysis of both 27 Y-STRs and 143 Y-SNPs in Eastern Han Chinese population.
Forens. Sci. Int. Genet. 2019,
42, e13–e20.
[Google Scholar]
-
39.
Pamjav H, Fothi A, Feher T, Fothi E. A study of the Bodrogkoz population in north-eastern Hungary by Y chromosomal haplotypes and haplogroups.
Mol. Genet. Genom. 2017,
292, 883–894.
[Google Scholar]
-
40.
Pankratov V, Litvinov S, Kassian A, Shulhin D, Tchebotarev L, Yunusbayev B, et al. East Eurasian ancestry in the middle of Europe: genetic footprints of Steppe nomads in the genomes of Belarusian Lipka Tatars.
Sci. Rep. 2016,
6, 30197.
[Google Scholar]
-
41.
Pimenoff VN, Comas D, Palo JU, Vershubsky G, Kozlov A, Sajantila A. Northwest Siberian Khanty and Mansi in the junction of West and East Eurasian gene pools as revealed by uniparental markers.
Eur. J. Hum. Genet. 2008,
16, 1254–1264.
[Google Scholar]
-
42.
Qi X, Cui C, Peng Y, Zhang X, Yang Z, Zhong H, et al. Genetic evidence of paleolithic colonization and neolithic expansion of modern humans on the tibetan plateau.
Mol. Biol. Evol. 2013,
30, 1761–1778.
[Google Scholar]
-
43.
Rowold DJ, Gayden T, Luis JR, Alfonso-Sanchez MA, Garcia-Bertrand R, Herrera RJ. Investigating the genetic diversity and affinities of historical populations of Tibet.
Gene 2019,
682, 81–91.
[Google Scholar]
-
44.
Song M, Wang Z, Zhang Y, Zhao C, Lang M, Xie M, et al. Forensic characteristics and phylogenetic analysis of both Y-STR and Y-SNP in the Li and Han ethnic groups from Hainan Island of China.
Forens. Sci. Int. Genet. 2019,
39, e14–e20.
[Google Scholar]
-
45.
Wang CC, Wang LX, Shrestha R, Zhang M, Huang XY, Hu K, et al. Genetic structure of Qiangic populations residing in the western Sichuan corridor.
PLoS ONE 2014,
9, e103772.
[Google Scholar]
-
46.
Wang M, He G, Zou X, Liu J, Ye Z, Ming T, et al. Genetic insights into the paternal admixture history of Chinese Mongolians via high-resolution customized Y-SNP SNaPshot panels.
Forens. Sci. Int. Genet. 2021,
54, 102565.
[Google Scholar]
-
47.
Wen SQ, Du PX, Sun C, Cui W, Xu YR, Meng HL, et al. Dual origins of the Northwest Chinese Kyrgyz: the admixture of Bronze age Siberian and Medieval Niru’un Mongolian Y chromosomes.
J. Hum. Genet. 2022,
67, 175–180.
[Google Scholar]
-
48.
Wen SQ, Sun C, Song DL, Huang YZ, Tong XZ, Meng HL, et al. Y-chromosome evidence confirmed the Kerei-Abakh origin of Aksay Kazakhs.
J. Hum. Genet. 2020,
65, 797–803.
[Google Scholar]
-
49.
Xie M, Song F, Li J, Lang M, Luo H, Wang Z, et al. Genetic substructure and forensic characteristics of Chinese Hui populations using 157 Y-SNPs and 27 Y-STRs.
Forens. Sci. Int. Genet. 2019,
41, 11–18.
[Google Scholar]
-
50.
Zhabagin M, Balanovska E, Sabitov Z, Kuznetsova M, Agdzhoyan A, Balaganskaya O, et al. The Connection of the Genetic, Cultural and Geographic Landscapes of Transoxiana.
Sci. Rep. 2017,
7, 3085.
[Google Scholar]
-
51.
Zhang D, Cao G, Xie M, Cui X, Xiao L, Tian C, et al. RETRACTED ARTICLE: Y Chromosomal STR haplotypes in Chinese Uyghur, Kazakh and Hui ethnic groups and genetic features of DYS448 null allele and DYS19 duplicated allele.
Int. J. Leg. Med. 2021,
135, 1119.
[Google Scholar]
-
52.
Zhang Y, Zhang R, Li M, Luo L, Zhang J, Ding J, et al. Genetic polymorphism of both 29 Y-STRs and 213 Y-SNPs in Han populations from Shandong Province, China.
Leg. Med. (Tokyo) 2020,
47, 101738.
[Google Scholar]
-
53.
Nei M, Tajima F. DNA polymorphism detectable by restriction endonucleases.
Genetics 1981,
97, 145–163.
[Google Scholar]