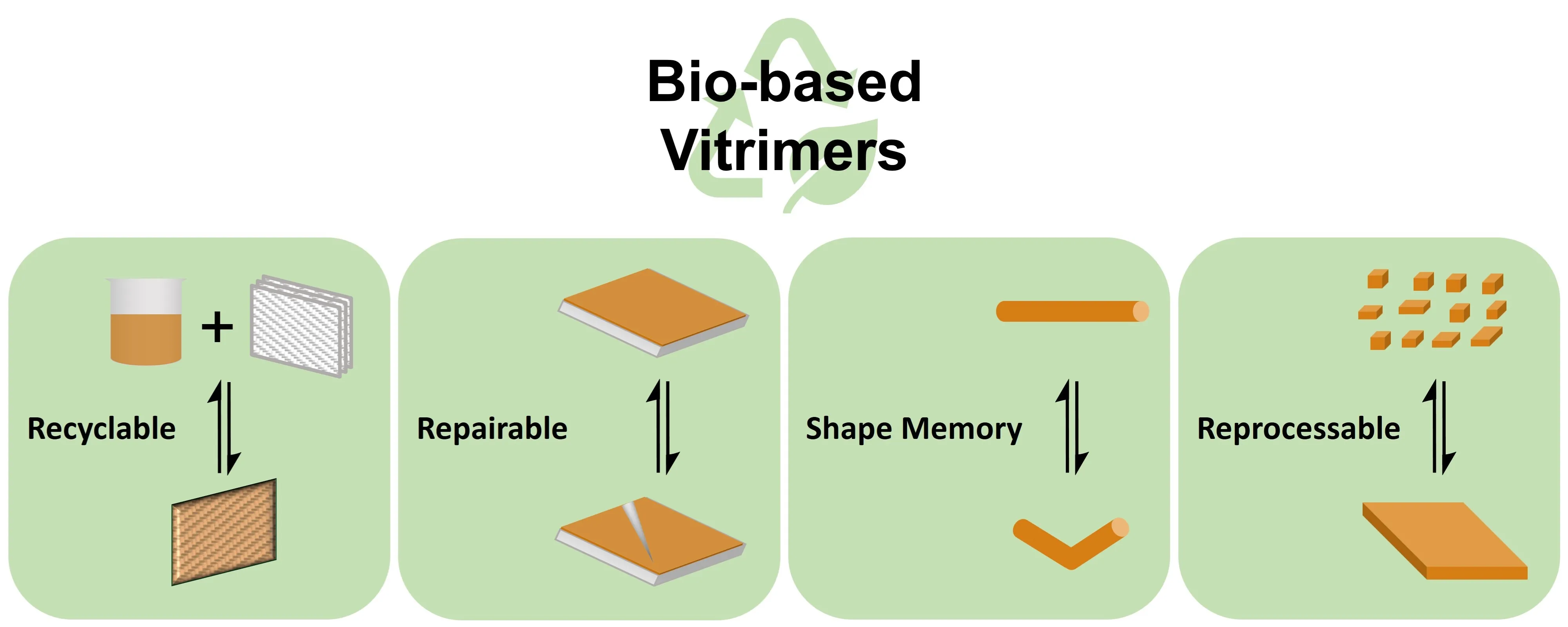
In 2011, Leibler and co-workers prepared the first vitrimer materials via curing epoxy resin with anhydride/fatty acids in the presence of zinc catalysts [
19]. Like thermosets, the prepared vitrimers have crosslinked network structures and thus have good chemical resistance. At elevated temperatures (>100 °C), the zinc salts promote dynamic transesterification reactions between ester bonds and hydroxyl groups, resulting in rearrangement of the crosslinked polymer network. As a result, the materials exhibit a certain degree of malleability, repairability, and recyclability, which are characteristic properties of thermoplastics. At lower temperatures, the exchange reactions are negligible and the materials have essentially fixed topology and behave like thermosets. Because vitrimer materials combine the advantages of both thermoplastics and thermosets, they have received extensive attention in recent years. Besides transesterification, other dynamic covalent bonds, such as imine, disulfide, and carbamate, have been used in vitrimer preparation [
20,
21].
2.1. Associative Networks vs. Dissociative Networks
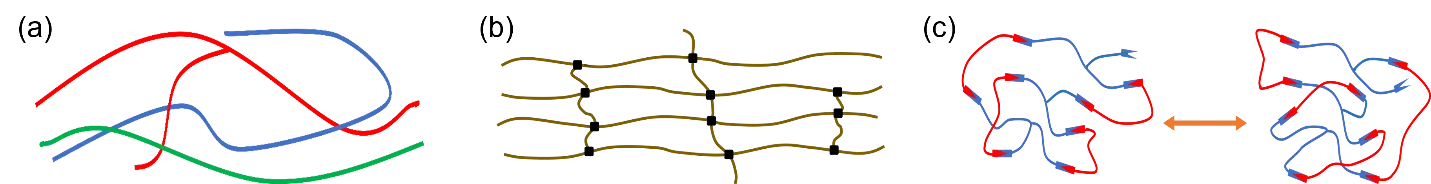
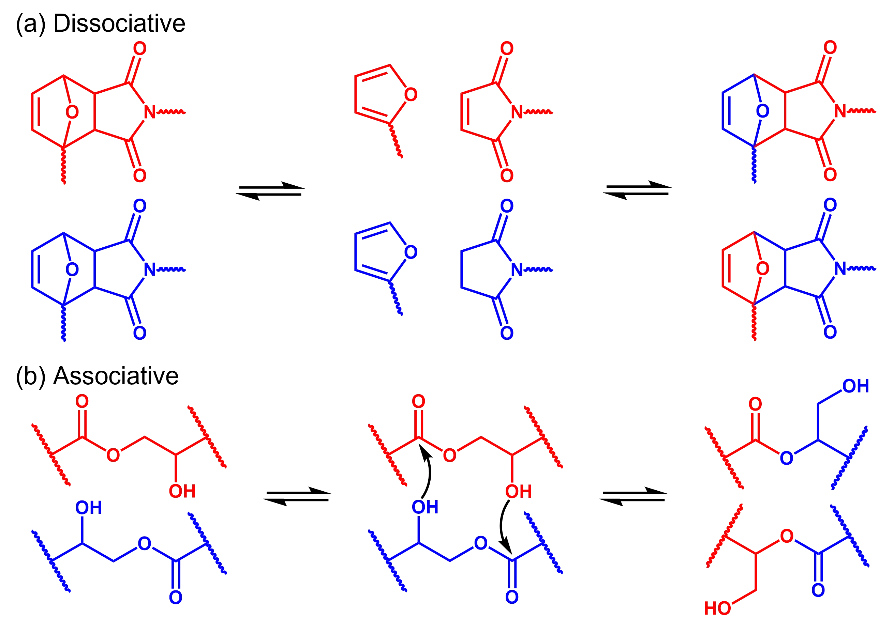
Not all crosslinked polymers containing dynamic covalent bonds are called vitrimers. According to the exchange mechanism, dynamic covalent polymer networks can be divided into two categories: dissociative networks and associative networks [
20]. In dissociative networks, cleavage of dynamic covalent bonds and rearrangement of the polymer network do not proceed simultaneously. For example, in polymers containing reversible furan-maleimide crosslinks, retro-Diels-Alder reaction is thermodynamically favorable at high temperatures while the cycloaddition reaction is favored at low temperatures. As a result, the furan-maleimide crosslinks break at elevated temperatures and reform when the material is cooled down (Scheme 1a). In associative networks, however, the bond exchange reactions and network rearrangement occur simultaneously (Scheme 1b). Therefore, the crosslink density of the polymer network stays constant, regardless of dynamic exchange reactions. Upon heating, these associative networks behave like vitreous silica and exhibit Arrhenius-like gradual viscosity decreases. Thus, Leibler and co-workers termed these associative networks as vitrimers [
19]. It is worth noting that several associative networks were reported before the vitrimer concept was introduced [
22,
23,
24,
25]. Also, several recent studies have shown that dissociative networks may have similar rheological behaviors as associated networks (vitrimers) [
26,
27]. Further, as detailed in Section 2.2 and Section 3, dynamic bonds in vitrimers could undergo dissociative reactions under specific conditions [
28].
Scheme 1. (<b>a</b>) Cleavage of dynamic covalent bonds and network rearrangement do not proceed simultaneously in dissociative networks. (<b>b</b>) Bond exchange reactions and network rearrangement occur simultaneously in associative networks.
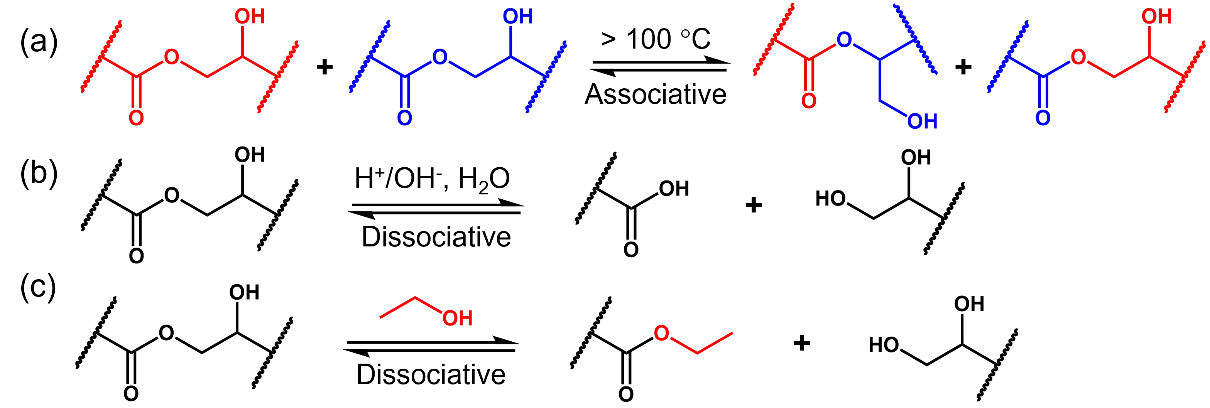
Under external stimuli, the associative bond exchange reactions in vitrimers enable rearrangement of the polymer network. Thus, vitrimers can exhibit certain degrees of processability and malleability without losing their network integrity. However, this solid-state plasticity of vitrimers is fundamentally different from the melt-flow behaviors of thermoplastics. Due to their crosslinked network structure and limited dynamic exchange reaction rates, most vitrimers cannot be reprocessed or recycled via injection molding or extrusion [
29]. In order to recycle these vitrimer materials, it is often necessary to decompose the polymer networks via dissociative bond cleavage of the dynamic crosslinks. For example, in vitrimers containing hydroxy-ester crosslinks, dynamic transesterification reactions between the esters and hydroxyl groups at high temperatures (>100 °C) enable network rearrangement and impart repairability, reprocessability, and shape changing properties to the materials (Scheme 2a). When end-of-life products need to be recycled, hydrolysis of the ester crosslinks in acidic or alkaline solutions could be used to disrupt the network structure (Scheme 2b). Alternatively, treatment in a large excess of alcohol also leads to decrosslinking of the polymer network (Scheme 2c). Notably, this process is reversible and the network could be reconstructed by removing the alcohol under heating [
30]. Similarly, depending on the conditions, both bond exchange and bond cleavage reactions could take place in other vitrimer systems, which will be discussed in Section 3.
Scheme 2. Associative bond exchange (<b>a</b>) and dissociative bond cleavage (<b>b</b>,<b>c</b>) reactions in hydroxy-ester vitrimers.
Stress relaxation is a typical feature of vitrimers. When a certain strain is applied to a vitrimer material, the generated internal stress will gradually decrease due to the network rearrangement caused by dynamic exchange reactions. Stress relaxation of vitrimer materials is a complex process and is related to many factors. Besides the type of exchange reactions, stress relaxation is also affected by the network crosslink density, the framework rigidity, polymer chain interactions (such as hydrogen bonding), and free volume in the material [
31,
32]. Because stress relaxation of vitrimers is enabled by exchange reactions, it is only effective when the temperature is above the glass transition temperature (
Tg) and the topology freezing transition temperature (
Tv). Below
Tg, the material would be in glassy state and the limited chain mobility restricts dynamic exchange reactions. Below
Tv, the kinetic of bond exchange reactions is too slow to induce topology change. Above both
Tg and
Tv, dynamic exchange reactions allow efficient stress relaxation, which can be accelerated by further increasing the temperature. The stress relaxation times at different temperatures follow the Arrhenius relationship and can be used to calculate the activation energy of dynamic exchange reactions [
33]. For vitreous silica,
Tv typically refers to the temperature at which the viscosity is ~10
12 Pa∙s [
34]. In vitrimers,
Tv can be determined by monitoring the dimensional change during heating of the material [
35,
36]. Alternatively,
Tv can be detected by fluorescence change of aggregation-induced-emission luminogens [
37].
The properties of many dynamic covalent bonds were recognized long before the vitrimer concept was introduced. Perhaps, early researchers focused on improving the stability and mechanical properties of materials and ignored the value of these dynamic covalent bonds in polymer recycling. Although many types of dynamic covalent crosslinks have been reported, the main ones used in bio-based vitrimers are hydroxy-esters, hydroxy-urethanes, imines, acetals, and disulfides.
3.1. Hydroxy-Esters
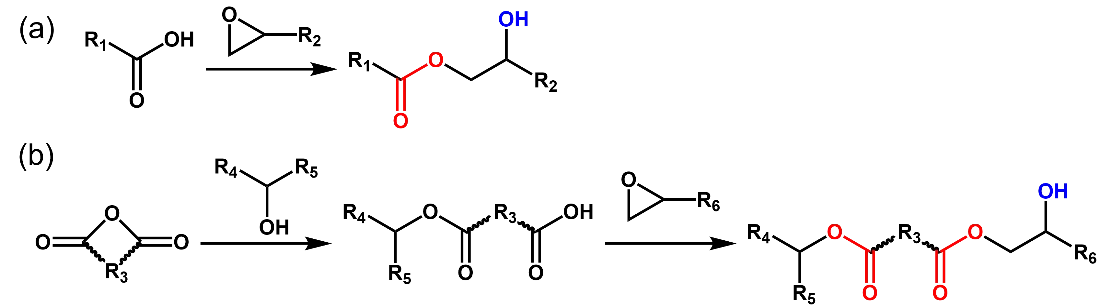
Hydroxy-ester vitrimers contain a large number of ester bonds and hydroxyl groups. Most hydroxy-ester linkages are prepared by the ring opening reaction of epoxies with carboxylic acids (Scheme 3a) or anhydrides (Scheme 3b). It is often necessary to add a certain amount of catalysts (>5 mol% of epoxy groups) to promote the curing reaction as well as transesterification. These catalysts mainly include Lewis acids (zinc acetate, zinc acetylacetonate, stannous octoate, etc.) [
38,
39] and tertiary amines (diazabicycloundecene (DBU), triazabicyclodecene (TBD), etc.) [
35,
40]. However, the addition of catalysts may lead to poor miscibility, corrosion of the substrate, and environmental issues from catalyst leaching [
21]. Recent studies demonstrated that catalyst-free hydroxy-ester vitrimers can be prepared via introducing excess hydroxy groups or monomers with tertiary amines into the polymer network [
41,
42,
43,
44,
45]. At present, renewable raw materials for hydroxy-ester vitrimers are mainly derived from vegetable oils, lignin, cellulose and natural rubber.
Scheme 3. Epoxy resin cured with carboxylic acid (<b>a</b>) and anhydride (<b>b</b>).
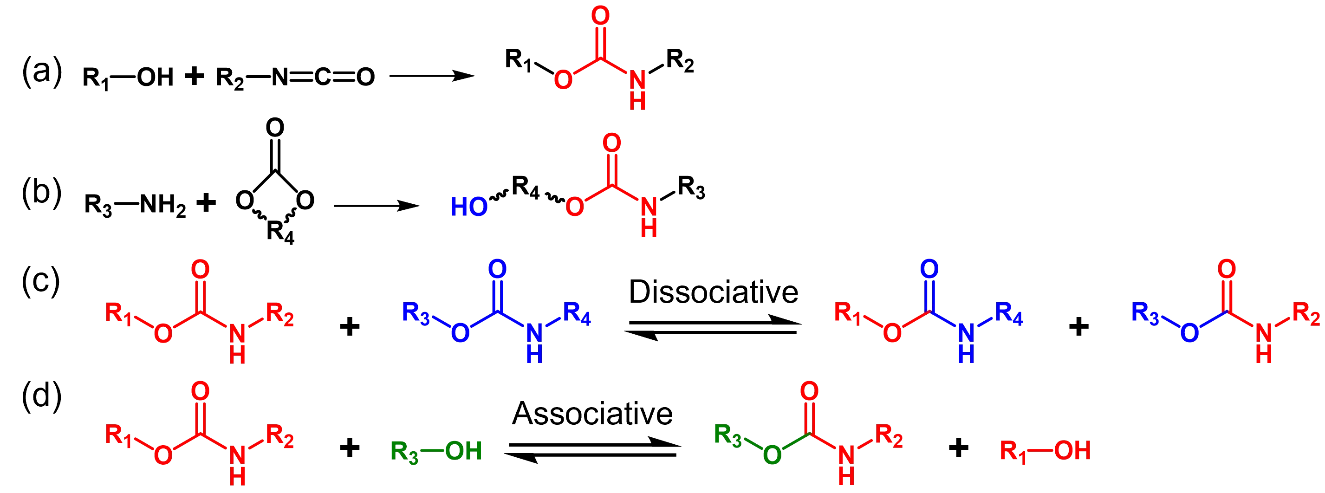
Hydroxy-urethane vitrimers are usually prepared via reaction of polyols with isocyanates (Scheme 4a). Alternatively, isocyanate-free polyhydroxyurethanes could be prepared from cyclic carbonates (Scheme 4b). Network rearrangement of hydroxyl-urethane vitrimers could be attributed to two factors: the dissociative carbamate exchange reaction (Scheme 4c) [
46] and the associative transcarbomylation reaction (Scheme 4d) [
47]. Even at high temperatures (>70 °C), carbamate exchange reactions require a certain amount of catalyst. Commonly used catalysts include dibutyltin dilaurate (DBTDL), bismuth neodecanoate, iron (III) acetylacetonate and other Lewis acids [
48,
49]. Similar to the hydroxy-ester system, introduction of excess hydroxyl groups or catalytic tertiary amines into the polymer network will result in catalyst-free hydroxy-urethane vitrimers [
47,
50]. Currently, biobased hydroxy-urethane vitrimers are mainly derived from vegetable oils.
Scheme 4. Preparation of carbamate linkage from isocyanate (<b>a</b>) and cyclic carbonate (<b>b</b>). (<b>c</b>) Dissociative carbamate exchange reaction. (<b>d</b>) Associative transcarbomylation reaction.
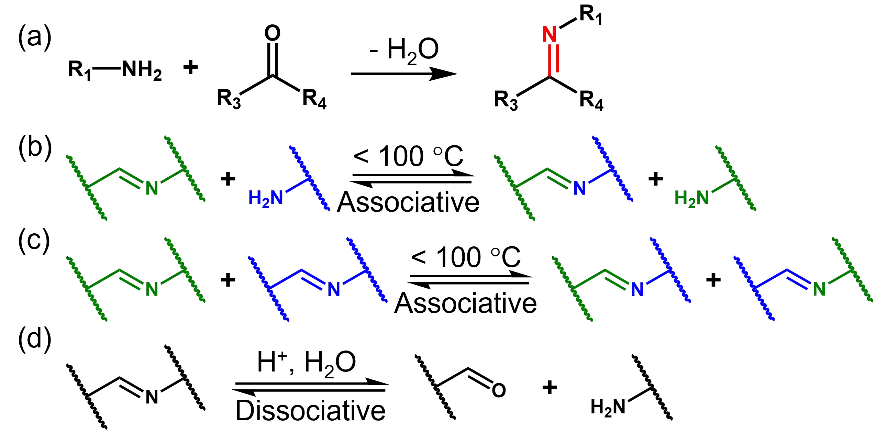
Imine linkages are formed by reacting aldehydes/ketones with amines (Scheme 5a). The formation of imines is reversible, and the by-product water must be continuously removed from the reaction system to achieve high yields [
51]. Compared with other dynamic bonds, imine formation takes place at mild conditions without catalysts and major side reactions [
52]. Imine exchange (Scheme 5b) and imine metathesis (Scheme 5c) endow polyimine vitrimers with stress relaxation, malleability, and self-healing properties [
20]. Compared with hydroxy-ester networks, the dynamic exchange reactions in polyimine vitrimers occur at lower temperatures and are more efficient. Moreover, these exchange reactions can be accelerated by adding excess amines [
53]. Imine linkages can be cleaved by acid hydrolysis (Scheme 5d), and thus polyimine vitrimers can be easily recycled. Due to the limited natural availability of aldehydes, most reported biobased polyimine vitrimers are derived from vanillin and its analogues.
Scheme 5. (a) Imine formation from ketones/aldehydres and amines. Imine exchange (<b>b</b>), metathesis (<b>c</b>) and hydrolysis (<b>d</b>).
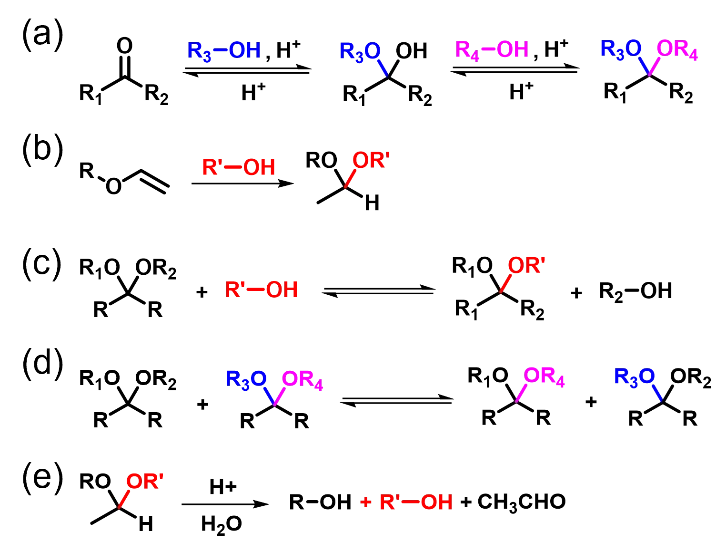
Acetals are commonly used as protecting groups for aldehydes and ketones in organic synthesis. Generally, there are two methods for constructing acetal linkages. In the first method, an acetal linkage is formed by the acid-catalyzed condensation reaction of a carbonyl group (from an aldehyde or ketone) with two hydroxyl groups (Scheme 6a) [
54]. In this approach, water is produced as a by-product and must be removed to push the reaction to completion. The two hydroxyls can be provided by separate alcohols or by a single diol or polyol. In the second method, the addition reaction of a vinyl ether with an alcohol yields the desired acetal linkage (Scheme 6b) [
55]. This addition reaction proceeds in one step without releasing small molecules, achieving a 100% atom economy. Scheme 6 shows the acetal exchange reactions via trans-acetalization (Scheme 6c) and acetal metathesis (Scheme 6d) [
28], as well as the acid-catalyzed hydrolysis of acetals (Scheme 6e). The acetal exchange reactions at high temperatures rearranges the polymer network, while acetal hydrolysis endows prepared materials with recyclability. Biomass resources for acetal dynamic covalent networks can be categorized into two groups: natural aldehydes like vanillin and natural polyphenols like lignin.
Scheme 6. (<b>a</b>) Acetal formation from aldehydes/ketones and alcohols. (<b>b</b>) Acetal formation from vinyl ethers and alcohols. (<b>c</b>) Trans-acetalization reaction. (<b>d</b>) Acetal metathesis. (<b>e</b>) Acid-catalyzed acetal hydrolysis.
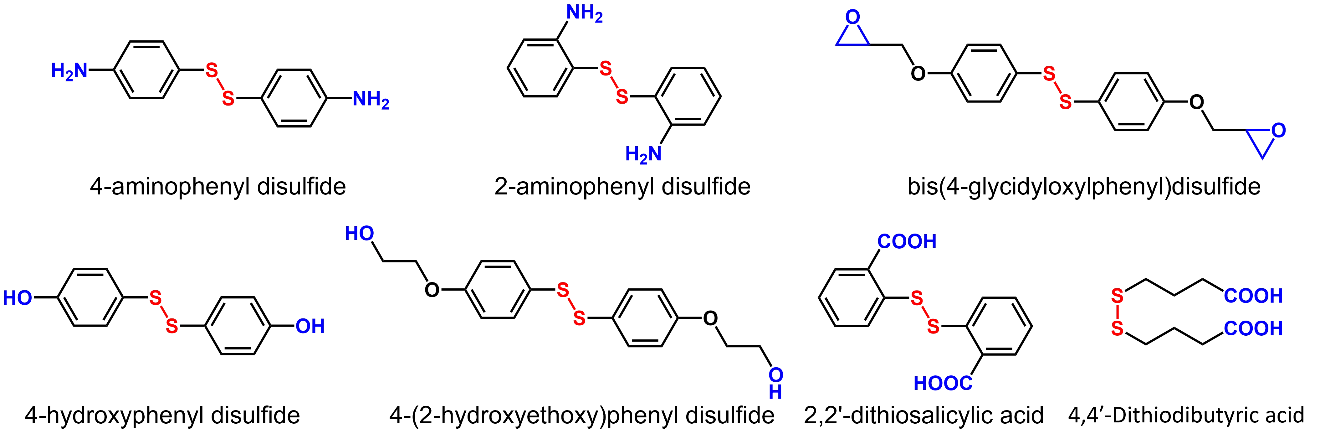
Vitrimers based on disulfide exchange are often prepared by introducing monomers containing disulfide bonds into the resin system. lists several commonly used disulfides [
56]. Disulfide exchange reactions could take place without a catalyst at high temperatures or under UV illumination. In crosslinked polymers containing thiuram disulfide moieties, the dynamic exchange reaction occurs even under ambient visible light at room temperature, enabling self-healing behavior in the bulk state [
57]. In addition to disulfide exchange reactions (Scheme 7a) that endow materials with typical vitrimer properties (self-healing, shape changing, reprocessing, etc.), disulfide bonds can also be decomposed by alkaline or reducing agents (Scheme 7b). As a result, vitrimers based on disulfide metathesis can be chemically degraded at room temperature into oligomers in aqueous solutions of sodium hydroxide or trialkylphosphines [
58,
59]. The thiols in the decomposition product can be oxidized to form new disulfide bonds. At present, biobased disulfide-containing vitrimers are mainly developed from natural rubber and vegetable oils.
. Disulfide-containing monomers commonly used in polymer preparation.
Scheme 7. Disulfide exchange (<b>a</b>) and disulfide decomposition (<b>b</b>) reactions.
Various biomass resources, including vegetable oils, fatty acids, lignins, natural rubber, cellulose, have been used as raw materials in vitrimer preparation. Compared to fossil resources, these biomass feedstocks are sustainable and environmentally friendly, which may offer a promising solution to plastic waste and pollution.
4.1. From Vegetable Oils and Fatty Acids
Vegetable oils and fatty acids have long been investigated as polymer precursors because they are widely available, relatively cheap, and intrinsically biodegradable. The double bonds on vegetable oils and fatty acids provide active sites for chemical modifications [
60]. They can be converted to various functional groups and used in vitrimer preparation, which will be discussed in detail below.
4.1.1. Hydroxy-Ester Vitrimers
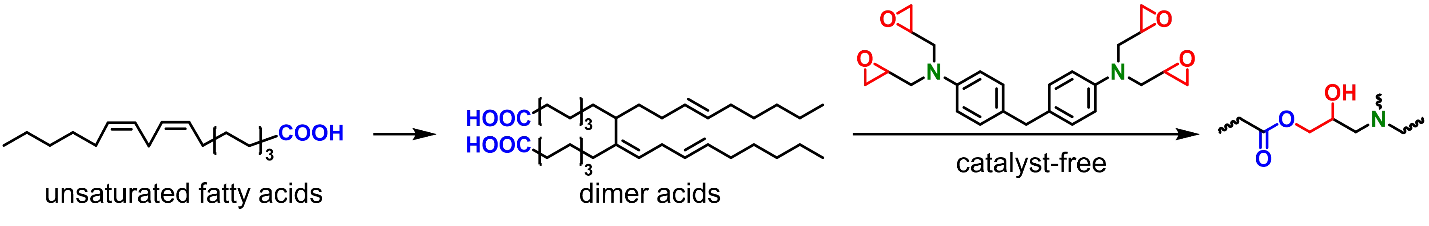
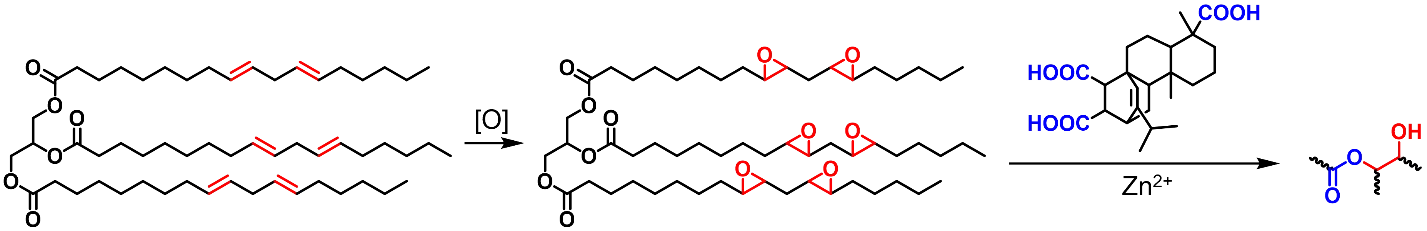
Vegetable oils and fatty acids could be modified in a few ways and used in hydroxy-ester vitrimers. Dimerization or trimerization of fatty acids through double bond addition reactions will produce polymeric fatty acids, which could be used as curing agents for epoxy vitrimers (Scheme 8). Vegetable oils could also be epoxidized and used as multifunctional epoxies (Scheme 9). For example, epoxidized soybean oil (ESO), a commercially available product with a well-established market, is often used as a multifunctional epoxy in vitrimer synthesis. Vegetable oils with higher degrees of unsaturation could be converted to polymer precursors via the Diels-Alder reaction (Scheme 10).
Polymeric Fatty Acids as Epoxy Curing Agents
The very first vitrimer prepared by Leibler and co-workers was obtained from thermal curing of diglycidyl ether of bisphenol A (DGEBA) with polymeric fatty acids in the presence of a zinc catalyst [
19]. The stoichiometric ratio of COOH to epoxy was set at 1:1, so abundant hydroxyl groups and ester linkages were preserved for transesterification. At this ratio, the bio-content of the prepared material was ~60 wt%. To ensure crosslinking, a mixture of trimer acid (77 wt%) and dimer acid (23 wt%) was used. The aliphatic chains of these polymeric fatty acids ensure the flexibility of the cured product. The tensile strength and elongation at break are 9 MPa and 180%, respectively. Under the catalysis of zinc ions, dynamic transesterification reactions are rapid because of the soft network. At 160 °C, the stress relaxation time is 1250 s. In comparison, the hard network prepared from glutaric anhydride and DGEBA exhibits a stress relaxation time of >10
4 s at 160 °C. Inspired by Leibler’s work, polymeric fatty acids have been used in the preparation of other hydroxy-ester vitrimers [
61,
62,
63,
64,
65].
Considering that zinc catalysts are corrosive and may harm the environment, our research group developed a catalyst-free vitrimer based on dimer acids and a glycidyl amine epoxy resin (Scheme 8) [
44]. Tetraglycidyl-4,4′-diaminodiphenylmethane (TGDDM) was selected as the glycidyl amine epoxy because it is widely used in the preparation of high-performance composites and adhesives. The tertiary amine groups of TGDDM act as internal catalysts for dynamic transesterification reactions and the stress relaxation time at 200 °C is ~7000 s. Therefore, the prepared material exhibits self-healing and shape-changing properties. When the molar ratio of epoxy to COOH is 1/1, the dimer acid content is ~70 wt%, and the tensile strength and elongation at break of the material are ~12.1 MPa and ~180%, respectively. This tensile performance is superior to most reported thermosetting elastomers based on fatty acids or vegetable oils. It is also found that the prepared elastomer could be decomposed in pure water when the temperature is above 170 °C. The obtained oligomers could be used to regenerate the thermosetting elastomer via hot pressing.
Scheme 8. Fatty adid-derived dimer acids can be used as epoxy curing agents.
Epoxidized Vegetable Oils as Multifunctional Epoxies
Aside from functioning as acid curing agents, vegetable oils can also be chemically modified with epoxy groups and used as epoxy resins. As early as 2013, Altuna et al. prepared a fully bio-based catalyst-free vitrimer from ESO and citric acid (CA) [
66]. When the molar ratio of epoxy to COOH was 2:1, the relaxation time of ESO-CA vitrimer at 160 °C was ~2 h. The low crosslink density of the cured product, the flexibility of the polymer chains, and the presence of abundant hydroxyl groups allow dynamic transesterification to occur without catalysts. On the other hand, these factors lead to low mechanical properties. The tensile strength of the prepared vitrimer material was only 0.5–0.6 MPa, which is far lower than commercial epoxies. Two recent studies replaced CA with more rigid glycyrrhizic acid (GL) and rosin-derived fumaropiramic acid (FPA) as crosslinkers for ESO (Scheme 9) [
67,
68]. The stress relaxation time of ESO-GL vitrimer was 1.14 h at 160 °C under TBD catalysis. Because of the transesterification reactions, ESO-GL vitrimer exhibited weldability, repairability, and shape memory properties. When used as recyclable adhesives, the lap-shear strengths were 3.42, 2.89, and 2.49 MPa for the original, repaired, and recycled samples [
67]. Because FPA has a more rigid skeleton than CA, ESO-FPA vitrimer exhibited much higher tensile strength (16 MPa) than that of ESO-CA vitrimer (0.5–0.6 MPa). However, the rigidity of the FPA moieties limits the kinetics of transesterification reactions. Even with the presence of zinc acetylacetonate (10 mol% of FPA), stress relaxation at 160 °C takes longer (~3 h) than catalyst-free ESO-CA vitrimer (~2 h) [
68]. Nicolas et al. epoxidized waste frying sunflower oil and cured with glutaric acid [
69]. Commercial petroleum-based di- and tri-epoxies were also added to improve the mechanical properties (tensile strength from 0.4 MPa to 17 MPa). In the presence of 1-methylimidazole, transesterification reactions enabled good shape memory behaviors.
Scheme 9. Epoxidized vegetable oils can be used as multifunctional epoxies.
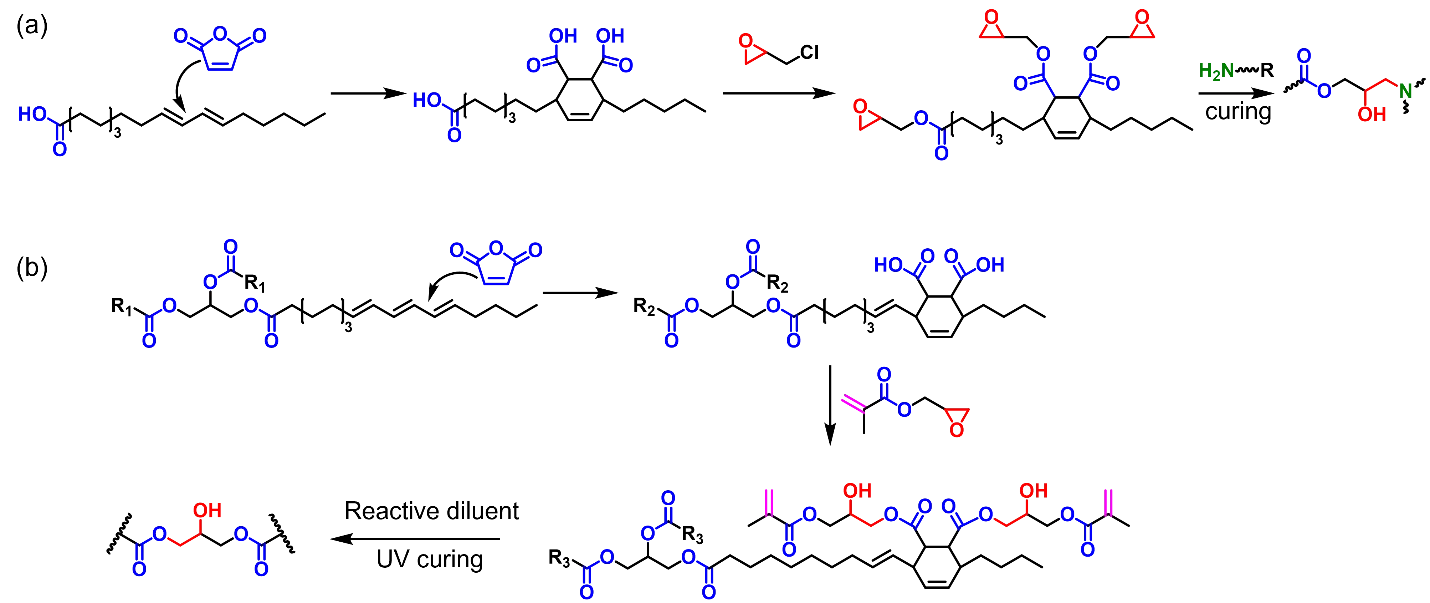
Diels-Alder Adducts of Vegetable Oils as Polymer Precursors
Vegetable oils with higher degrees of unsaturation could undergo Diels-Alder reaction to prepare polymer precursors with reactive functional groups. Following this method, our research group prepared a hempseed oil epoxy (HOEP) with glycidyl ester moieties and mixed it with DGEBA to prepare room-temperature curable coatings (Scheme 10a) [
70]. During the curing process, triethanolamine (TEOA) was used as a co-curing agent to introduce tertiary amines and excess hydroxyl groups into the polymer network. The reaction between glycidyl ester epoxy and amine curing agent yield β-hydroxy esters. TEOA catalyzes the transesterification reaction between β-hydroxy esters and excess hydroxyls, endowing the coatings with repairability at high temperatures. To prepare ambient pressure recyclable vitrimers from HOEP, we further incorporated a dialdehyde into the HOEP-amine system [
71]. Because amines can simultaneously react with both epoxy and aldehyde groups [
72], the resulting vitrimers possess not only ester but also imine dynamic linkages. The low viscosities of the resins and the intrinsic recyclability of the resulting dual dynamic network vitrimers make them ideal for recyclable composite applications. Zhang et al. prepared a UV-curable oligomer from tung oil (TO) and cured it with a malic acid-based reactive diluent under UV radiation (Scheme 10b) [
73]. Both the oligomer and the reactive diluent possess multiple hydroxyl and ester groups, facilitating dynamic transesterification reactions at high temperatures. When recycled via hot pressing, further polymerization occurred and the recycled material exhibited improved tensile strength and modulus.
Scheme 10. Vegetable oils with higher degrees of unsaturation could be modified via Diels-Alder reaction to prepare multifunctional glycidyl ester epoxies (<b>a</b>) or UV curable methacrylates (<b>b</b>).
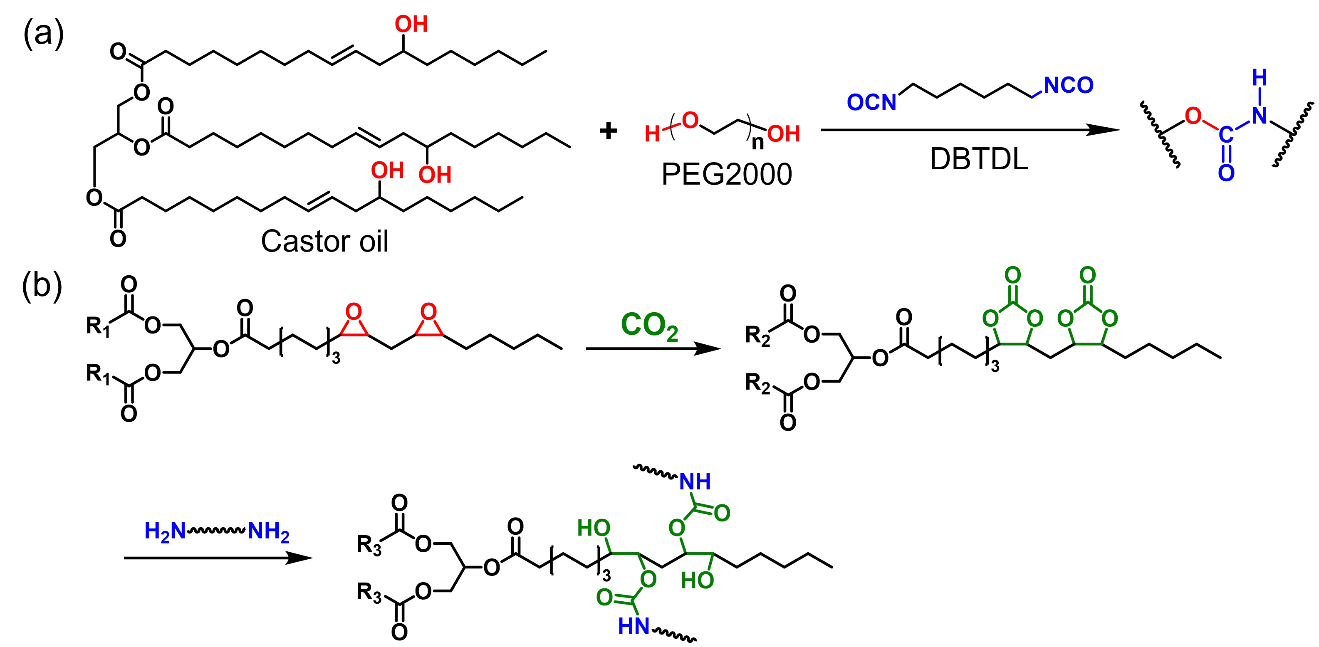
4.1.2. Hydroxy-Urethane Vitrimers
The hydroxyl groups in castor oil make it an appealing raw material in polyurethane (PU) synthesis. However, castor oil derived thermosetting PUs are non-reprocessable. In this context, Yan et al. used castor oil, polyethylene glycol, and hexamethylene diisocyanate (HDI) as raw materials, DBTDL as catalyst, and carbon nanotubes (CNTs) as functional filler and prepared PU vitrimer nanocomposites (Scheme 11a) [
74]. Under high temperature (>90 °C) and DBTDL catalysis, carbamate exchange reactions endow the materials with weldability and shape memory properties. Taking advantage of the photothermal effect of CNTs, the weldability and shape memory properties of the vitrimer nanocomposites can also be excited by light irradiation. Shi et al. prepared PU prepolymers with isocyanate end groups from castor oil, 1-decanol, and isophorone diisocyanate (IPDI) and cured with bisphenols to prepare reprocessable crosslinked Pus [
75]. The kinetics of network rearrangement can be regulated via tuning the electron-withdrawing ability of bisphenols. The prepared PU elastomers have tensile strengths greater than 5 MPa and elongations at break greater than 300%. Under the catalysis of 1,4-diazabicyclo[2.2.2]octane, the materials could be reprocessed via hot pressing and the mechanical properties of reprocessed samples are similar to those of the original one. Instead of using isocyanates like HDI and IPDI, Liu et al. prepared fully biobased polyhydroxyurethanes from cyclic-carbonated soybean oil and 1,8-diamino-p-menthane (Scheme 11b) [
76]. Due to the presence of large amounts of hydroxyls, transcarbomylation reactions occur in the absence of catalysts and provide the material with shape memory, self-healing, reprocessing and recycling properties. After three cycles of remolding, the material recovered > 85% of its original strength (~2 MPa).
Scheme 11. (<b>a</b>) Preparation of PU from castor oil and isocanates. (<b>b</b>) Preparation of vegetable oil-derived isocyanate-free polyhydroxyurethane.
4.1.3. Disulfide Vitrimers
When disulfide-containing curing agents are incorporated into vegetable oil-based elastomers, reprocessable elastomers are obtained. Chen et al. first prepared PU precursors from castor oil and excess HDI, and then cured them with 4-aminophenyl disulfide [
77]. This disulfide-containing monomer not only introduces dynamic crosslinks into the network but also brings in hydrogen bonding. As a result, the prepared polyurethane-urea exhibits good mechanical properties, with a tensile strength and elongation at break of 9.4 MPa and 188%, respectively. Moreover, due to disulfide exchange reactions, the elastomer can be reprocessed via compression molding. After three cycles of reprocessing, the sample shows mechanical properties comparable to those of the original sample, with a tensile strength of ~ 9.0 MPa and an elongation at break of ~ 185%. In another report, the same group prepared poly(ester amide) vitrimers from sebacic acid, castor oil, 4-aminophenyl disulfide and polyamide 1010 monomer salt via amidation and esterification [
78]. The higher the 4-aminophenyl disulfide content, the faster the relaxation. When the 4-aminophenyl disulfide content is higher than 8 wt%, the materials exhibit excellent malleability and can be reprocessed by compression molding at 200 °C under 10 MPa for only 10 min. The mechanical properties stay about the same after several cycles of thermal remolding, with tensile strengths of ~12 MPa and elongations at break of ~300%.
4.2. From Lignins
Given its abundance and rigid aromatic structures, lignin is potentially an ideal feedstock for renewable polymer production. Lignin has abundant aliphatic and aromatic hydroxyls and thus can be chemically modified and used in polymer preparation.
4.2.1. Hydroxy-Ester Vitrimers
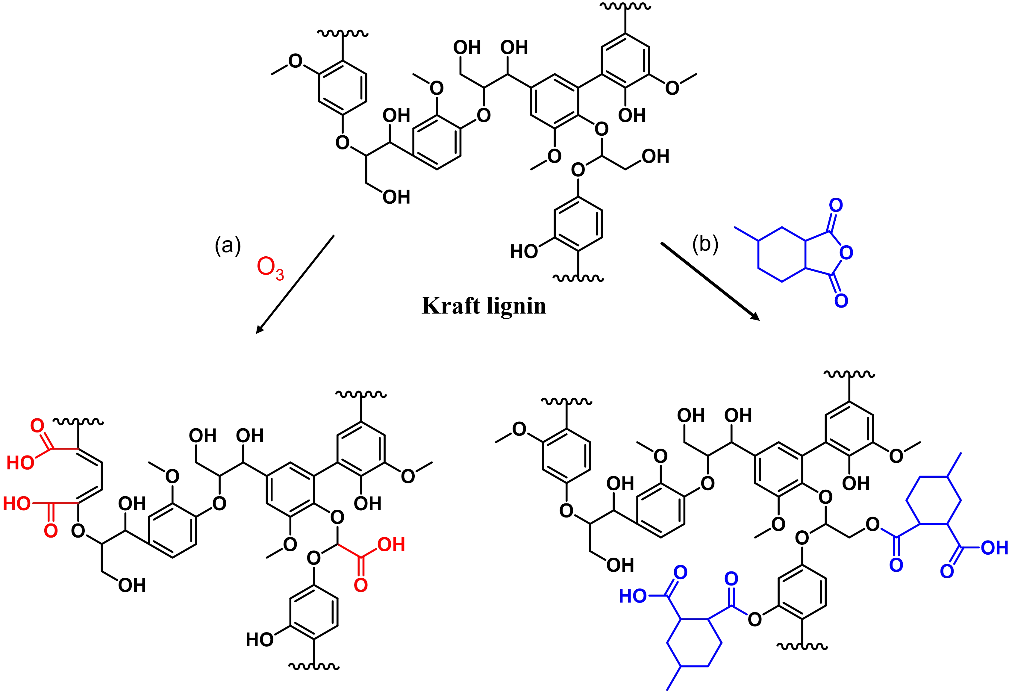
The hydroxyl groups of lignin can be chemically modified into carboxylic acids via oxidation or reaction with an anhydride (Scheme 12). The obtained lignin carboxylic acids can be used as curing agents for epoxy resins. Alternatively, both aliphatic and aromatic hydroxyls of lignin can be reacted with epichlorohydrin to yield epoxidized ligin. When such epoxidized lignin is cured with an acid, a lignin-based hydroxy-ester vitrimer is obtained (Scheme 13).
Modified Lignin as Epoxy Curing Agents
Our research group used two different treatment methods to prepare lignin carboxylic acids (Scheme 12) [
79,
80]. In the first study, Kraft-lignin (KL) was treated with ozone to generate abundant carboxyl groups. After oxidation, a certain portion of the aromatic rings were cleaved and opened, thus reducing the stiffness of lignin. The reduction of rigid ring structures and the increase in carboxyl content improved the miscibility of ozonated KL with epoxy resins. When cured with diglycidyl ester of sebacic acid, a fully biobased vitrimer with high lignin content was prepared. With the presence of a zinc catalyst and excess phenolic hydroxyls, the relaxation time of the prepared lignin vitrimer was only 81 s at 200 °C. When used as adhesive to bond metal sheets, the lignin vitrimer exhibited cohesive failure behavior with a lap shear strength of 6.5 MPa. Moreover, the failed two halves could be re-bonded via dynamic transesterification reactions. The recovered adhesive still showed excellent strength, with a shear stress of 5.0 MPa. In our second study, KL was chemically modified with an anhydride to yield lignin with adequate carboxyl groups. The modified KL was then employed as a curing agent for poly(ethylene glycol) diglycidyl ether. The cured product showed good adhesion and hardness properties when used as coatings on tin plates. More importantly, the lignin coating can be removed via soaking in dilute sodium hydroxide (NaOH) solution at room temperature for 10 min. During soaking, the excess phenolic hydroxyls react with NaOH to form hydrophilic phenate ions, which cause the coating to swell and lose its adhesion properties. In comparison, conventional thermosetting coatings cannot be easily removed without damaging the substrate. Further, due to the rapid transesterification reactions, the lignin coatings are repairable with the assistance of ethylene glycol.
Scheme 12. Chemical modification of Kraft lignin with ozone (<b>a</b>) or anhydride (<b>b</b>) enables lignin as epoxy curing agents.
Modified Lignin as Epoxy Resin
Epoxidized enzymatic hydrolysis lignin was also used to prepare hydroxy-ester vitrimers (Scheme 13). Xue et al. mixed epoxidized enzymatic hydrolysis lignin with DGEBA in different proportions and cured with dodecanedioic acid to prepare lignin vitrimers [
81]. The mechanical properties of the obtained vitrimer materials could be adjusted by varying the lignin content. When using epoxidized enzymatic hydrolysis lignin alone (without DGEBA), the sample exhibits a high tensile strength of 39.5 MPa. In the presence of zinc acetylacetonate hydrate, all lignin vitrimers exhibit excellent shape memory, self-healing and reprocessing properties. Similarly, Tang et al. prepared fully bio-based lignin vitrimers from epoxidized fractionated lignin and sebacic acid [
82]. Enzymatic hydrolysis lignin was sequentially extracted with different organic solvents and then epoxidized with epichlorohydrin. The epoxidized fractionated lignin was then cured with sebacic acid under Zn
2+ catalysis. The material prepared from epoxidized ethanol-soluble lignin reports a high tensile strength of 39.82 MPa. This is due to the lower molecular weight of ethanol-soluble lignin and the higher crosslink density resulting from higher epoxy content. The authors noted that increasing phenolic hydroxyl content facilitated the self-repairing performance of the lignin vitrimers.
Scheme 13. Epoxidized enzymatic hydrolysis lignin can be cured with acids to prepare hydroxy-ester vitrimers.
4.2.2. Acetal Networks
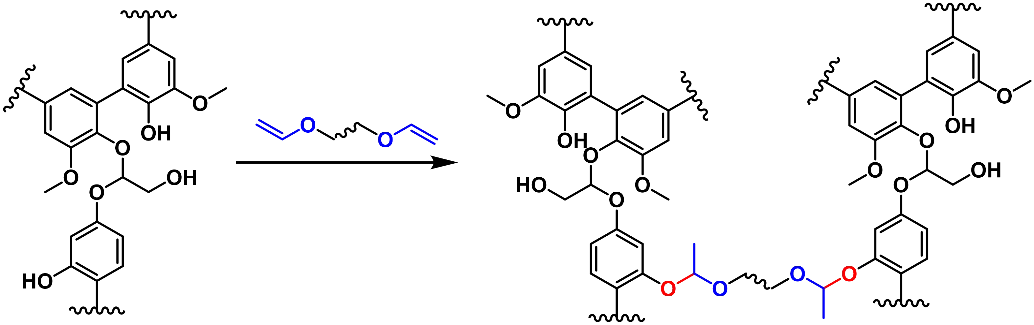
Lignin as a polyphenol can also be reacted with vinyl ethers to construct acetal networks (Scheme 6b). Using this approach, Moreno et al. prepared recoverable adhesives via reacting softwood kraft lignin with poly(ethylene glycol) divinyl ether (Scheme 14) [
83]. Due to the rapid acetal exchange reactions, the prepared lignin vitrimer could be reprocessed at 150 °C via compression molding. When used as an adhesive for aluminum, the sample exhibited a lap sheer strength of 6.0 MPa. When the failed two halves were re-glued via hot pressing at 150 °C, 93% of the adhesion performance (5.6 MPa) was recovered.
Scheme 14. Preparation of acetal networks from lignin and vinyl ether.
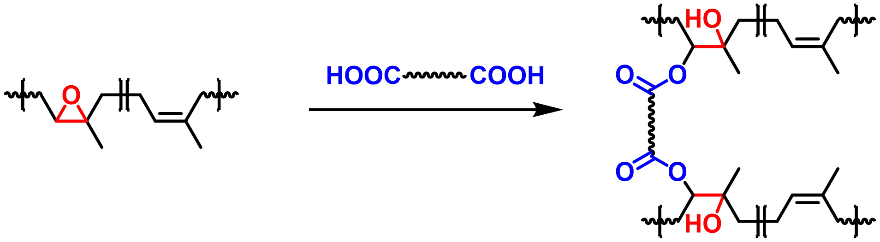
Natural rubber is produced from rubber trees grown in the tropics and accounts for nearly half of all rubber products on the market (tires, shoe soles, gaskets, etc.). Conventionally, natural rubber is crosslinked via vulcanization, which forms a permanent network structure and improves the material’s chemical resistance and elasticity. Because of the stable crosslinks, conventional natural rubbers cannot be reprocessed. To prepare vitrimer materials from natural rubber, epoxidized natural rubber (ENR) is often cured with acid crosslinkers (Scheme 15). Alternatively, disulfide bonds could be introduced into the rubber network via using disulfide-containing curing agents or changing the sulfur content during vulcanization.
4.3.1. Hydroxy-Ester Vitrimers
In 2016, Imbernon et al. cured ENR with dodecanedioic acid under the catalysis of zinc acetate and imidazole (Scheme 15) [
84]. To obtain a rubber material with low crosslink density and excellent elasticity, only 3.2 parts of dodecanedioic acid were added to 100 parts of ENR. The prepared rubber material exhibits obvious stress relaxation and two rubber sheets could be adhered together via compression at 200 °C, indicating that dynamic transesterification occurs and leads to network rearrangement. Inspired by this work, a series of studies on the preparation of ENR vitrimers using diacids or polyacids as crosslinkers were reported. The acid crosslinkers include carboxyl-modified carbon black [
85], carboxyl-modified carbon nanodots [
86], carboxyl-modified cellulose nanocrystals [
87], carboxyl-modified chitosan [
88], etc. The carboxyl groups react with epoxies on ENR to form β-hydroxy ester linkages, which undergo transesterification reactions at high temperatures. These carboxyl-modified crosslinkers also act as reinforcing agents. Thus, the modulus and strength of the material increase with increasing crosslinker content. However, at higher crosslinker content, the chain mobility of the polymer network is restricted, which in turn limits the rate of transesterification reactions. Instead of using direct thermal energy for transesterification reactions, Feng et al. prepared an ENR vitrimer with photothermal agent aniline trimer [
89]. Under light irradiation, the aniline trimer moieties convert light energy into heat, which raises the temperature of the material and induces dynamic exchange reactions. As a result, the shape memory and self-healing properties can be triggered via light illumination. Liu et al. reported a method to effectively enhance the mechanical properties of ENR vitrimer without sacrificing the reprocessability by introducing hydrogen bonds [
90]. ENR was crosslinked by sebacic acid and grafted with N-acetylglycine simultaneously. N-acetylglycine introduces hydrogen bonding into the crosslinked network, thereby improving the modulus and tensile strength of the ENR vitrimer. More importantly, the incorporation of hydrogen bonding also facilitates the network rearrangement. After three generations of recycling, the ENR vitrimer retained more than 80% of its original mechanical properties.
Scheme 15. Crosslinking of epoxidized natural rubber (ENR) with acid crosslinkers.
4.3.2. Disulfide Vitrimers
In industrial production, natural rubber is often crosslinked by sulfur during vulcanization. The network after conventional vulcanization consists of monosulfide (0–5%), disulfide (30–40%), and polysulfide (60–70%) crosslinks [
91]. In the absence of catalysts, only disulfide bonds can undergo dynamic exchange reactions. Due to their limited content of disulfide bonds, traditional rubber products cannot be reprocessed or repaired. A dynamic network with repairability and reprocessability could be prepared via increasing the disulfide content. Hernández et al. demonstrated that the disulfide/polysulfide ratio in crosslinked natural rubber could be adjusted by changing the sulfur content [
92]. When the mass ratio of natural rubber to sulfur is 100/0.7 (CV
1), 100/1.3 (CV
2) and 100/2.5 (CV
3), the disulfide/polysulfide ratios in the vulcanized rubbers are 0.48, 0.35, and 0.32, respectively. These results show that reduction in sulfur content leads to an increased disulfide/polysulfide ratio. The resulting rubbers with more disulfide linkages (CV
1 and CV
2) showed a higher healing capability compared to the one with the least disulfide bonds (CV
3). The repairability of CV
1 and CV
2 systems was attributed to the dynamic exchange reactions of disulfide linkages. It is worth noting that the reduction of sulfur content also leads to lower crosslink density and poorer mechanical performance. In comparison, the tensile strength of CV
3 is ~6 MPa, while that of CV
1 and CV
2 is only ~0.5 MPa. The lower crosslink density leads to better chain mobility, which also contributes to the improved healing capability [
93].
Disulfide bonds can also be introduced into the rubber network via using disulfide-containing curing agents. Imbernon et al. prepared a rubber vitrimer by curing ENR with 4,4′-dithiodibutyric acid, a disulfide-containing diacid [
94]. The metathesis of disulfide bonds at high temperatures (180 °C) rearranges the polymer network and thus the rubber material shows fast stress relaxation and good adhesion behavior. Moreover, grinded samples could be recycled via compression molding and regenerated sample retained ~ 50% of the initial strength. Compared with the hydroxy-ester ENR vitrimer prepared by the same group [
84], the disulfide ENR vitrimer exhibits faster stress relaxation and does not require catalysts. The stress relaxation time of the disulfide ENR vitrimer is less than 0.5 h at 180 °C, while that of the hydroxy-ester ENR vitrimer is ~4 h at 200 °C. In both studies, the authors mentioned the possibility of double bond oxidation and self-polymerization at high temperatures. These side reactions may deteriorate the mechanical properties of reprocessed samples. Cheng et al. cured ENR with two aromatic disulfides and prepared recyclable rubber materials [
58]. When 2,2′-dithiosalicylic acid is used alone as the crosslinker, the resulting material has a high tensile strength (10.8 MPa) yet the healing efficiency is low (64%). When 4-aminophenyl disulfide is used as the only crosslinker, the healing efficiency was higher at 79%, yet the tensile strength is only 4.6 MPa. When both aromatic disulfides are used, the obtained rubber material showed an excellent healing efficiency of 98% with good mechanical strength (9.3 MPa). It is believed that the hydrogen bonding between amine moieties and ester groups contributes to the improvement in mechanical strength and healing efficiency.
4.4. From Polysaccharides
Polysaccharides like cellulose and chitin possess abundant hydroxyl groups. These hydroxyl groups can participate in dynamic transesterification reactions. Further, these hydroxyl groups could be converted to carboxyl groups and used to cure epoxy resins (Scheme 16).
Cellulose is the most abundant organic polymer in nature and is widely used in paper and textile industries. Several studies have investigated the use of cellulose in vitrimer preparation. Lossada et al. prepared nanopapers from cellulose nanofibers (CNF) [
95]. First, vitrimer nanoparticles were synthesized from emulsion polymerization of epoxy resin and fatty acid. Then, TEMPO-treated nanocellulose and synthesized vitrimer nanoparticles were dispersed in water to form a suspension. After water evaporation, the obtained nanocellulose/vitrimer films underwent dynamic transesterification at 120 °C and formed transparent nanopapers. The mass fraction of cellulose in the prepared nanopapers is greater than or equal to 50 wt%. The hydrophobic vitrimer nanoparticles improved the water resistance and ductility of the nanocomposites. After soaking in water for 4 h, the tensile strength and elongation at the break of the nanopaper were ~30 MPa and ~26%, respectively. Moreover, two nanopapers can be welded together by dynamic transesterification at 120 °C. In another report, Xu et al. first hydrolyzed and oxidized cotton in a hydrochloric acid/nitric acid mixture to yield carboxylated cellulose nanocrystals (CCNs). The prepared CCNs with a large number of carboxyl groups were then reacted with DGEBA to produce healable and recyclable composites [
96]. Similarly, Li et al. used carboxymethyl cellulose (CMC) as a crosslinker and reacted with DGEBA in water [
97]. After evaporation of water and subsequent thermal curing, a CMC-based vitrimer was prepared. With the help of glycerol and TBD catalyst, good reprocessability was obtained.
Scheme 16. The hydroxyl groups of cellulose could be converted to carboxyl groups and used as crosslinkers for epoxy resins.
Similar to cellulose, chitin possesses abundant hydroxyl groups and thus exhibits good interfacial interactions with hydroxy-ester vitrimers. Accordingly, Chen et al. prepared a layered composite using chitin as filler and hydroxy-ester vitrimer as matrix polymer [
98]. Chitin films were first prepared with the help of ionic liquid, and then impregnated with vitrimer resin. After curing, a weldable and reprocessable chitin-vitrimer composite was obtained. When the composite is welded with the neat vitrimer, the obtained bilayer structure could be used as a smart actuator because the two layers respond asymmetrically to thermal stimuli.
4.5. From Natural Aldehydes
Biobased aldehydes including vanillin and furfural derivatives have been used in constructing polyimine and acetal networks. Imine likages are prepared via reacting aldehydes with amines (Scheme 5a) and acetal linkages are formed when aldehydes are reacted with alcohols (Scheme 6a).
4.5.1. Polyimine Vitrimers
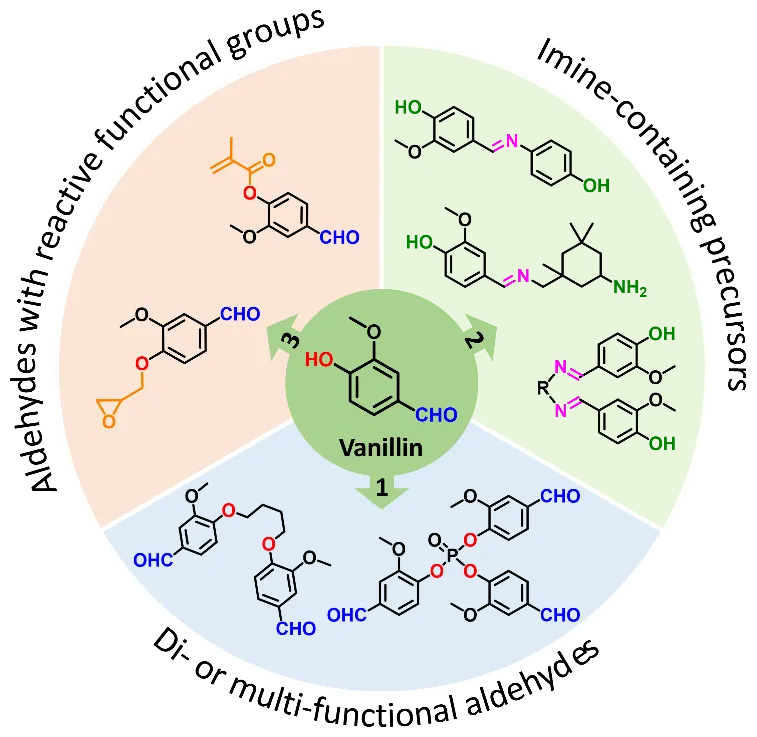
Due to the limited natural availability of aldehydes, most reported biobased polyimine vitrimers are derived from vanillin and its analogues. Vanillin is a phenolic aldehyde found in vanilla bean and can be produced on large scale from lignin [
99]. In general, vanillin can be functionalized in three ways to prepare polyimine networks (): (1) Generate di- or multi-functional aldehydes from vanillin, and then react with di- or polyamines; (2) Synthesize imine-containing precursors from vanillin, then use the precursors as epoxy curing agents or for further functionalization; (3) Convert the phenolic hydroxyl of vanillin into reactive functional groups, and then construct polymer networks using the reactive functional group and the remaining aldehyde.
. Functionalization of vanillin into precursors for polyimine networks.
Following the first method, several di- or multi-functional aldehydes have been synthesized and used in vitrimer preparation. For example, Geng et. al. prepared a di-aldehyde from vanillin via Williamson ether synthesis using 1,4-dibromo butane as the organohalide [
100]. This di-aldehyde was then crosslinked by commercial amines to form reprocessable and recyclable polymer networks. Similarly, Wang et. al. and Liu et. al. synthesized tri-functional and multi-functional aldehydes from vanillin, respectively [
101,
102]. Polyimine vitrimers derived from these aldehydes presented excellent mechanical and thermal properties because of their abundant aromatic rings. Moreover, the phosphorus content and aromatic rings in the obtained materials contributed to their excellent flame retardancy. Song et al. cured the tri-functional aldehyde with soybean oil-derived polyamines to produce reusable adhesives. The prepared polymers have both dynamic imine bonds and hydrogen bonds, making them sensitive to temperature changes. Moreover, the mechanical strength of the materials could be tuned by varying the imine content [
103]. Other methods are used to synthesize di- and tri-aldehydes as well. In Hong et al.’s work, vanillin is converted into a dialdehyde via the Duff reaction [
104]. In Zhang et al.’s work, vanillin was first reacted with epichlorohydrin to give an epoxy precursor, which was then reacted with a tri-thiol to yield a tri-aldehyde [
105]. It is worth mentioning that all the above di- or multi-functional aldehydes are solids and therefore require solvents in polymer preparation.
The second method involves the synthesis of imine-containing precursors from vanillin. Following this approach, Liu et. al. first synthesized a diphenol from vanillin and aminophenol and then used it to cure a biobased epoxy resin (glycerol triglycidyl ether) [
106]. The obtained vitrimer has a Young’s modulus of 1.6 GPa and a tensile strength of 62 MPa, which are close to those of commercial epoxies. Xu et al. converted the above diphenol into a diepoxy via reaction with epichlorohydrin and cured with 4,4-diaminodiphenylmethane (DDM) [
107]. The obtained epoxy vitrimer shows excellent thermal and mechanical properties, with a tensile strength of 93 MPa, a Young’s modulus of 2.2 GPa, and a
Tg of 181 °C. These values are even higher than those of commercial epoxy thermosets. The authors attributed the excellent performance to the conjugated structure between the imine bond and the neighboring benzene rings. Vanillin-derived phenols containing imine linkages could also be synthesized using different diamines. When used as epoxy curing agents, the resulting vitrimers exhibit satisfactory tensile properties and excellent solvent resistance [
108,
109,
110].
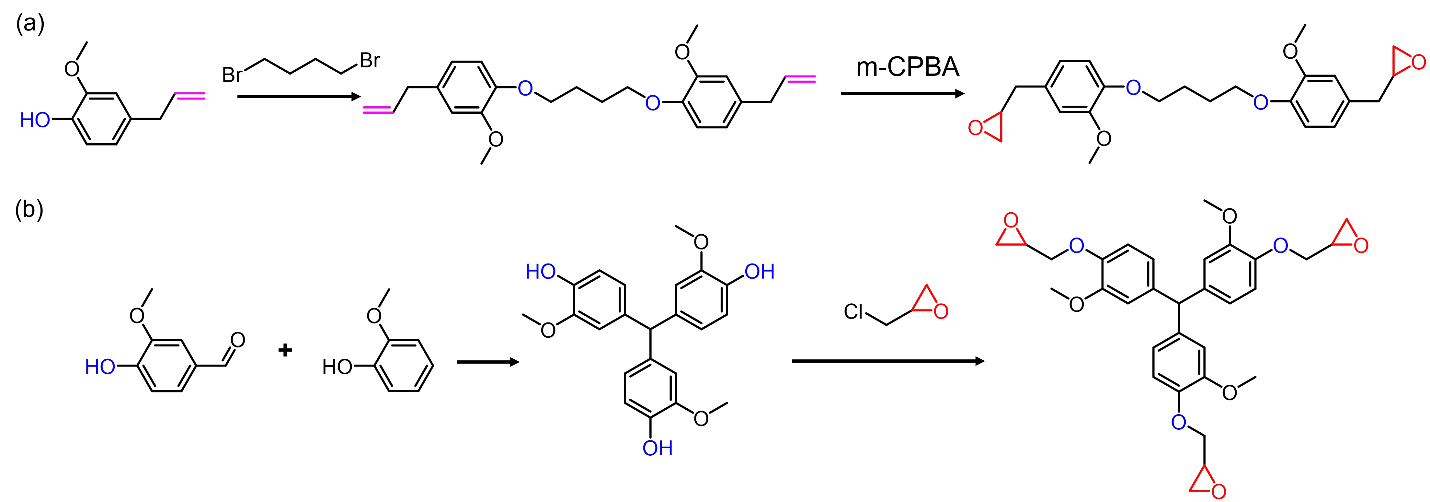
In the third method, the phenolic hydroxyl of vanillin is first converted into an epoxy or methacrylate, the epoxy/methacrylate group together with the remaining aldehyde are then used to build the dynamic network. Wang et al. synthesized vanillin monoepoxide via reacting vanillin with epichlorohydrin and cured with a cyclic diamine [
111]. During curing, the epoxide and aldehyde can simultaneously react with the diamine curing agent. The obtained vitrimer possessed comparable properties to commercial epoxy resins. At high temperatures (>160 °C), rapid imine exchange reactions rearrange the polymer network, and thus the material could be reprocessed within 20 min at 180 °C under 15 MPa. During reprocessing, the mechanical properties of the sample are well-reserved. Similarly, Su et al. prepared a diepoxy from a vanillin derivative with two phenolic hydroxyls and cured with a diamine [
112]. Because both epoxy groups and aldehyde groups could act as crosslinking sites, the prepared vitrimer has higher crosslink density and better thermal-mechanical performance than the DGEBA counterpart. Snyder et al. incorporated vanillin monoepoxide into a polyester backbone via ring-opening copolymerization of cyclic anhydride and epoxide, and then crosslinked it with diamines [
113]. The rheological behaviors and crosslink densities of the obtained vitrimers can be easily manipulated by varying the epoxide co-monomer and its content. Besides vanillin-based epoxy precursors, vanillin-based methacrylate was also synthesized and used to construct polyimine networks. Xu et al. reacted methacrylated vanillin with di- and tri-amines and prepared imine-containing di- and tri-methacrylates, which can be photocured to produce chemically recyclable and thermally reprocessable polyimine networks [
114]. Gao et al. used methacrylated vanillin and lauryl methacrylate to modify lignin via reversible addition-fragmentation chain transfer polymerization [
115]. The aldehyde groups (from vanillin moieties) in the grafted lignin were then crosslinked by diamines to produce dynamic networks, which can be used as self-healing adhesives.
In addition to vanillin, other natural aldehydes have also been employed in preparing polyimine vitrimers. Since syringaldehyde has a structure similar to vanillin, it can be functionalized in a similar manner and used to construct polyimine networks. Xie et al. synthesized a fully biobased imine-containing diphenol by reacting syringaldehyde with 1,4-butanediamine [
116]. The obtained diphenol was then reacted with epichlorohydrin to yield a diepoxy, which was cured with DDM to produce a vitrimer with excellent flame retardancy and high mechanical properties. Moreover, the incorporation of imine bonds makes the material chemically degradable. Dhers et al. developed a fully biobased polyimine vitrimer from 2,5-furandicarboxaldehyde and Priamine 1071 [
117]. In order to accelerate the imine exchange reactions, excess amines were introduced into the system. The obtained material exhibits fast stress relaxation at low temperatures (1300 s at 40 °C). However, the long chain structure of Priamine 1071 greatly limits the materials’ mechanical properties (with a tensile strength of 0.7 MPa and an elongation at break of 24%).
4.5.2. Acetal Networks
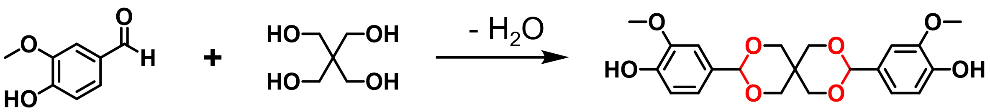
Biobased aldehydes can also be reacted with alcohols to construct acetal networks (Scheme 6a). In 2019, Ma et al. prepared an epoxy resin with spiro diacetal structure from vanillin and used it to prepare recyclable carbon-fiber-reinforced polymer (CFRP) composites and removable coatings [
118]. In this study, two vanillin molecules were linked by pentaerythritol to produce a diphenol with spiro diacetal structure (Scheme 17). The diphenol was then reacted with epichlorohydrin to yield a solid diepoxy. After curing with isophorondiamine (IPDA) with the help of a solvent, the obtained material exhibited mechanical properties comparable to those of commercial epoxy resins. Moreover, the acetal linkages are readily degradable in mild acidic conditions, which ensures the recyclability of the prepared material. Inspired by this work, Wang et al. incorporated the same spiro diacetal structure into polybenzoxazines and prepared recyclable yet high-performance CFRPs [
119]. Notably, in both studies, solvents are required in CFRP fabrication because the acetal precursors are solids. Accordingly, Li et al. synthesized a liquid diepoxy with acetal moiety from vanillin and trimethylolpropane [
120]. The obtained diepoxy has a viscosity of 7.3 Pa∙s at 30 °C and thus can be used to fabricate CFRP via vacuum-assisted resin transfer molding. Miao et al. synthesized a dimethacrylate from the above diphenol with spiro diacetal structure and used it as a crosslinker in photo-curing 3D printing [
121]. When mono-functional 4-acryloylmorpholine was used as the reactive diluent, printed structures can be dissolved in acidic solutions and used as sacrificial molds. Li et al. prepared an acetal precursor from furfural and produced reprocessable polyurethanes (PU) [
122]. To adjust the flexibility of the PU material, bismaleimide was added to form dissociative Diels-Alder bonds with the furan ring.
Scheme 17. Synthesis of acetal-containing precursor from vanillin and polyol.
Natural polyols such as glycerol and erythritol could be used together with vanillin to prepare acetal networks with high bio-content. For example, Wang et al. synthesized a diol with acetal moiety from vanillin and glycerol. The fully biobased diol was then used to replace bisphenol A in epoxy and PU preparation [
123,
124]. Similarly, Yuan et al. prepared a diepoxy with dicyclo diacetal structure from vanillin and erythritol [
125]. Owing to their rigid ring structures, these biobased acetal networks present excellent mechanical and thermal properties. Kirchhecker et al. synthesized an acetal diol from 5-hydroxymethylfurfural and glycerol and used it in the preparation of recyclable PU adhesives [
126].
4.6. From Other Biomass Feedstocks
Given the diversity of biomass feedstocks, other biomass resources have also been used in preparation of vitrimer materials.
4.6.1. Hydroxy Ester Vitrimers
Our research group prepared eugenol-based (Scheme 18a) and guaiacol-based (Scheme 18b) epoxy resins and reacted them with anhydride curing agents to prepare vitrimers, respectively [
36,
127]. Notably, guaiacol-based tri-epoxy was synthesized via reacting the phenolic hydroxyls with epichlorohydrin. Because of its rigid structure, the prepared vitrimer material exhibits a
Tg value greater than 170 °C and a tensile strength of ~60 MPa. These properties are comparable to those of commercial high-performance epoxies. Similarly, Oh et al. converted the phenolic hydroxyls of naringenin into epoxides and cured it with a diacid to prepare a recyclable vitrimer with shape-memory behavior.[
128] Liu et al. developed catalyst-free vitrimers from itaconic acid, glycerol, and maleic anhydride [
129]. The prepared material exhibited good malleability and was used to fabricate CFRPs. The obtained CFRP could be recycled by treating with 1M NaOH aqueous solution. Brutman et al. prepared vitrimers from polylactic acid (PLA) [
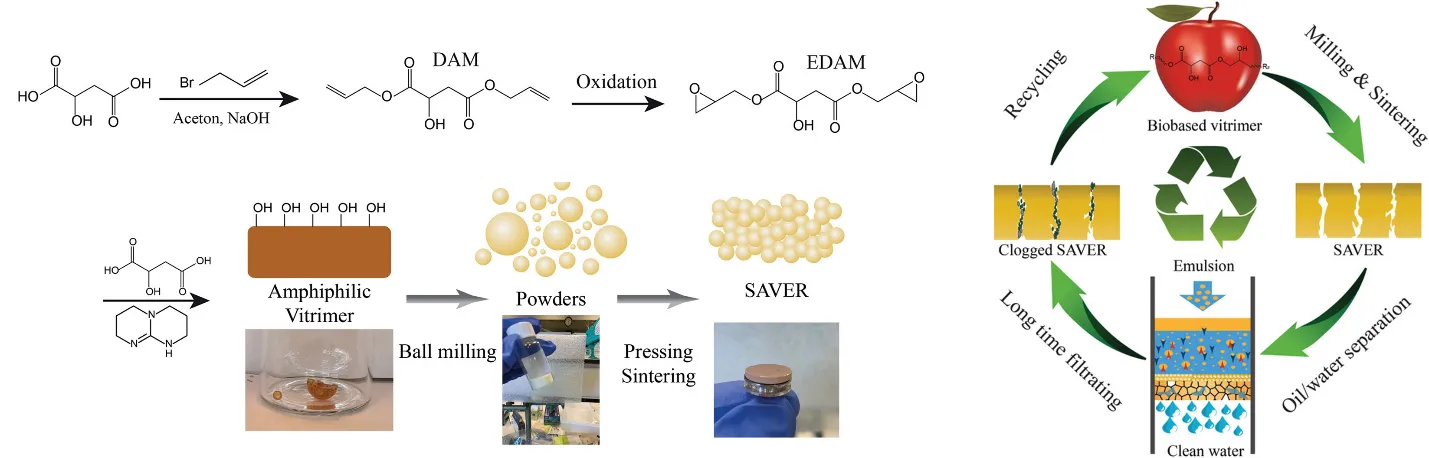
130]. First, they prepared a multi-arm PLA with hydroxyls as end groups. Then, the PLA was reacted with methylenediphenyl diisocyanate under the catalysis of stannous octoate to form vitrimers, which exhibited good reprocessability. Ye et al. prepared a recyclable superamphiphilic vitrimer from malic acid and used it for efficient oil-water separation [
131]. When the superamphiphilic membrane is irreversibly blocked, it can be depolymerized into soluble oligomers in ethylene glycol. After filtering off the insoluble contaminants, the superamphiphilic vitrimer could be reproduced via evaporating the ethylene glycol, thus achieving a closed-loop life cycle (). Li et al. reacted the phenolic hydroxyls of tannic acid with maleic anhydride to prepare polycarboxylic acids and used them to cure epoxidized vegetable oils [
132]. The resulting vitrimers exhibited excellent thermal stability and can be mechanically recycled.
Scheme 18. Two different methods of preparing biobased epoxies: (<b>a</b>) from epoxidation of double bonds and (<b>b</b>) by reacting hydroxyl or carboxyl groups with epichlorohydrin.
. Biobased amphiphilic vitrimer with a closed-loop life cycle and its use in oil-water separation. (Reprinted with permission from Reference [
131]; Copyright (2021) John Wiley and Sons).
4.6.2. Acetal Networks
Natural polyphenols could be reacted with vinyl ethers to prepare acetal networks (Scheme 6b). Using this method, Li et al. crosslinked epigallocatechin gallate with tri(ethylene glycol) divinyl ether and prepared weldable and extrudable materials [
133]. The fast reprocessability originates from accelerated acetal exchange reactions by neighboring phenolic hydroxyls. Moreover, the high rigidity of the biobased polyphenol and the high crosslink density provide high mechanical performance.
4.6.3. Disulfide Vitrimers
Ma et al. cured isosorbide-derived epoxy with 4-aminophenyl disulfide and obtained an epoxy vitrimer with disulfide linkages [
59]. Although the mechanical properties are slightly lower than the counterpart cured by traditional curing agent 4,4′-methylenedianiline, the prepared vitrimer exhibits reprocessability, degradability and shape memory behavior. Lee et al. synthesized a curing agent containing both imine and disulfide moieties from cystine and vanillin (Scheme 19) [
134]. The carboxylic acid and phenolic hydroxyl of this fully biobased curing agent can both react with isocyanates to produce crosslinked PU materials. Due to the synergistic effects of imine exchange and disulfide exchange, the PU materials have excellent recyclability and repairability.
Scheme 19. Synthesis of a fully biobased curing agent containing both imine and disulfide moieties.
This research was funded by United States Department of Agriculture, National Institute of Food and Agriculture (USDA NIFA) (Award No. 2020-67021-31138).
Reference collection, B.Z., T.L. and M.F.; Visualization, B.Z., Y.C. and M.F.; Writing—Original Draft Preparation, B.Z., M.F., C.H. and T.L.; Writing—Review & Editing, B.Z., B.B. and J.Z.; Funding Acquisition, T.L. and J.Z.
Not applicable.
Not applicable.
This research was funded by United States Department of Agriculture, National Institute of Food and Agriculture (USDA NIFA) (Award No. 2020-67021-31138).
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.