Lignocellulose is an abundant biomass feedstock that can be converted to fuels and high-value chemicals [
1,
2,
3,
4,
5]. Lignin is one of three main components of lignocellulose and has great potential utility for the production of value-added chemicals and biofuels [
5,
6,
7,
8,
9]. There are three important types of ether C–O bonds in lignocellulose, either β-O-4, α-O-4, and 4-O-5 linkages, where the effective transformation of lignin compounds cannot be efficiently achieved using conventional chemical processes due to the inactive nature of these ether C–O bonds [
10]. The ether cleavage problem has been extensively studied with many catalyst materials [
10,
11,
12]. Ni-based catalysts are frequently used for the reductive cleavage of these linkages in conventional thermal processing. An unfortunate consequence of the harsh reaction conditions required to activate the stable C–O linkages in these processing routes, such as high reaction temperature and high hydrogen pressure, is that they cause side reactions to occur, while the process is also energy-intensive.
The localized surface plasmon resonance (LSPR) effect of the plasmonic metals (gold, silver, copper) NPs has recently been utilized in developing new photocatalysis processes under moderate reaction conditions [
13,
14,
15,
16,
17]. The enhanced electromagnetic (EM) near-field of the irradiated plasmonic metal NPs and the light excitation of hot electrons are two of the main physical effects that drive chemical reactions with light in the presence of these catalyst NPs [
18,
19,
20,
21]. We previously reported that for the C–O bond cleavage of benzyl phenyl ether, the light irradiation of supported Ag and Au NPs significantly improved the catalytic activity of Ni
2+ complexes that had previously been immobilized onto an inert support [
18]. A careful study of the reaction mechanism revealed that light-excited hot electrons transferred from the plasmonic metal NPs to the immobilized Ni
2+ ions and reduced them to Ni
0 atoms that behaved as active sites that were able to cleave ether C–O linkages. Analysis of electron paramagnetic resonance (EPR) data revealed that light-excited hot electrons transferred from the illuminated plasmonic NPs to the immobilized Ni
2+ ions in the presence of aromatic molecules, molecules possessing conjugated π-orbitals [
18]. The catalysts were prepared by immobilizing Ni
2+ ions onto a metal oxide support by grafting silane with amino-linker groups, and this system exhibited high activity toward α-O-4 linkage cleavage but poor performance toward the cleavage of robust 4-O-5 linkage in similar model substrates.
In the present study, photocatalysts containing Ni
2+ were prepared using a simpler and more practical approach: Ni(OH)
2 was formed in situ under basic conditions from Ni(NO
3)
2 and KOH. Ni(OH)
2 precipitates were attached to the plasmonic metal Au NPs hosted on ZrO
2 support. ZrO
2 is inert support for catalytic reactions and does not absorb visible light, and it has the advantage that metal NPs tend to bind strongly to its surface, compared to silica or alumina [
13,
14]. After the overall catalyst structure was deposited, photoexcited electrons at the Au NPs could transfer to Ni(OH)
2, reduce the Ni
2+ and generate active catalytic Ni
0 species on the outmost surface of the catalyst. The reactant molecules with aromatic ether C–O bonds could readily access these catalytic sites, leading to the cleavage of the ether C–O bonds in the reaction. Furthermore, the reaction was active when nickel(II) acetylacetonate [Ni(acac)
2], nickel(II) chloride [NiCl
2] and nickel(II) acetate [Ni(OAc)
2] were employed as a Ni
2+ source. These observations provided additional proof that the illuminated Au NPs can promote the catalysis of Ni(II) species from different nickel salts. This novel Au/ZrO
2–Ni(OH)
2 catalyst system achieved high conversion of the model aromatic ether compounds, under visible light irradiation, at low reaction temperatures approaching 40 °C, which is much lower than the optimized reaction temperature (90 °C) reported previously for a similar photocatalyst system [
18]. This Au/ZrO
2–Ni(OH)
2 catalyst system also remained stable after the fifth cycle in catalyst recycling studies.
*2.1. Preparation of Catalysts*
2.1.1. ZrO
2 Supported Au Nanoparticles (Au/ZrO
2)
The catalysts with gold weight percentages of 0.5%, 1%, 3%, 5% and 10% were prepared by the impregnation-reduction method [
13]. For example, 1 wt% Au/ZrO
2 was prepared by the following procedure: 2.0 g commercially available ZrO
2 powder (particles size less than 100 nm) was dispersed into 10.12 mL of 0.01 M HAuCl4 aqueous solution was added while magnetically stirring. 5.3 mL of 0.1 M lysine was then added to the mixture with vigorous stirring for 30 min to adjust the solution pH to 8–9. To this suspension, 2 mL of 0.35 M NaBH
4 solution was added dropwise in 5 min. The mixture was then aged overnight, and then the solid was separated, washed with water for three times and ethanol once, and then dried in a vacuum oven at 60 °C. The dried solid was used directly as the catalyst. Catalysts with other ratios were prepared in a similar method but using different quantities of HAuCl
4, lysine and NaBH
4.
2.1.2. ZrO
2 Supported Ni NPs (Ni/ZrO
2)
Catalysts of 5 wt% Ni were prepared by an impregnation-reduction method similar to Au/ZrO
2. The only difference is using 85 mL 0.01 M NiCl
2 aqueous solution with 1.0 g ZrO
2 support.
2.1.3. Reduction of Ni(II) Species in H
2 Atmosphere
As-prepared catalyst Ni/ZrO
2 above was reduced in a tube furnace and reduced by hydrogen reduction under 5% H
2 and Ar gas mixture atmosphere at 600 °C for 1 h.
2.1.4. Au/ZrO
2–Ni(OH)
2 Catalyst
Au/ZrO
2–Ni(OH)
2 was prepared during the reaction. For example, 75 mg Au/ZrO
2 was added to 0.05 mmol Benzyl Phenyl Ether (α-O-4), 0.05 mmol KOH and 2 mL isopropyl alcohol solution in the 10 mL glass tube. Then 0.02 mmol Ni(NO
3)
2·6H
2O was added to the solution to form Ni(OH)
2 by reacting with KOH. Au/ZrO
2–Ni(OH)
2 can work together directly as a photocatalyst.
*2.2. Photocatalytic Activity Test*
In a typical activity test, a 10 mL reaction tube was used as the reactor. After 0.05 mmol reactant, 0.05 mmol KOH and 75 mg catalyst had been loaded, 0.02 mmol Ni(NO
3)
2·6H
2O was added to the solution to form Ni(OH)
2 by reacting with KOH. Argon was bubbled into the reaction solution for 1 min, and the reaction tube was sealed to isolate the reaction from the air. The reaction tube was then stirred magnetically and irradiated with a halogen lamp (from Nelson, 500 W, wavelength in 400–750 nm range; VIC, Australia). The irradiance was set to certain intensity (e.g., 0.5 W/cm
2) unless otherwise specified. The reaction temperature was monitored using a thermal camera (FLIR One Pro; QLD, Australia). Without reaction temperature control, light irradiation caused the reaction temperature to increase rapidly. The reactant vessel was therefore cooled to control the reaction temperatures < 70 °C. A heater was used to set reaction temperatures when temperatures > 70 °C were investigated. The control reaction in the dark was conducted using an oil bath placed above a magnetic stirrer, and the reaction tube was wrapped with aluminum foil to isolate the contents from stray light. The temperature of dark reactions was maintained at the same temperature as the corresponding reaction performed under irradiation. At the end of each reaction time, 1 mL aliquots were collected and filtered through a Millipore filter (pore size of 0.45 μm) to remove the particulate matter. The clear liquid-phase products were analyzed with an Agilent 6980 gas chromatography (GC) (Agilent, Santa Clara, CA, USA) using an HP-5 column to analyze the change in the concentrations of reactants and products. An Agilent HP5973 (Agilent) mass spectrometer was used to identify the products.
Action spectrum experiments were conducted with light emitting diode (LED) lamps (Tongyifang, Shenzhen, China) with wavelengths of 395 ± 5 nm, 450 ± 5 nm, 520 ± 5 nm, 590 ± 5 nm, and 620 ± 5 nm. The light intensity at the reaction vial was measured to be 0.2 W/cm
2 using an AULTT energy meter (model: CEL-NP2000), and other reaction conditions were kept identical to those of typical reaction procedures. The apparent quantum yield (AQY) was calculated as follows: apparent quantum yield = [(*M*
light − *M*
dark)/*N*
p] × 100%, where *M*
light and *M*
dark are the molecules of products formed under irradiation and dark conditions, respectively, *M* = mole number of the reactant × conversion × 6.02 × 1023 (Avogadro constant). *N*
p is the number of photons involved in the reaction. *N*
p = *E*
total/*E*
1, *E*
total (the total energy involved in the reaction irradiation) = intensity × light spot area×reaction time, *E*
1 (the energy of one photon) = h × c/λ (h is Planck constant, c is light speed, and λ is the wavelength of the LED light).
*2.3. Catalyst Characterization*
The morphology and elemental composition of photocatalysts were studied using a JEOL 2100 transmission electron microscopy (TEM) equipped with a Gatan Orius SC1000 CCD camera, energy dispersion X-ray (EDX) spectrometer (X-MAXN 80TLE, OXFORD Instruments; JEOL, NSW, Australia) was coupled for elemental analysis. The accelerating voltage of TEM was 200 kV. Diffuse reflectance UV-visible spectra of the catalysts were collected with a Cary 5000 UV-Vis-NIR spectrometer from Agilent company (Santa Clara, CA, USA) using BaSO
4 as a blank reference. The scanning scope was 200 nm to 800 nm. X-ray diffraction (XRD) patterns were recorded on a Philips PANalytical X’ Pert PRO diffractometer using Cu Kα radiation (λ = 1.5418 A); the fixed power source was 40 kV and 40 mA. The diffraction data were collected from 5° to 75° at a scanning rate of 2.5°/min with a resolution of 0.01°. X-ray photoelectron spectroscopy (XPS) analysis was performed with a Kratos Axis Ultra photoelectron spectrometer using mono Al Kα (1486.6 eV) X-ray. Inductively coupled plasma-atomic emission spectroscopy (ICP-AES) analysis was performed using a Perkin Elmer 8300 DV ICP (PerkinElmer, MA, USA) fitted with an ESI SC-4DX auto-sampler and PrepFAST 2 sample handling unit for online internal reference and auto-dilution of samples and calibration references. Nitric acid, purified by sub-boiling distillation, was used for the preparation of all references and blank solutions were used throughout the analysis. X-band (ca. 9.77 GHz) continuous-wave (CW) EPR spectra were recorded on a Bruker Biospin Elexsys E500 EPR spectrometer (Bruker, VIC, Australia) fitted with a super-high-Q cavity (CW EPR). Spectra were recorded under the following conditions: room temperature, modulation frequency 100 kHz, modulation amplitude 0.1 mT, and microwave power 20 mW (10 dB).
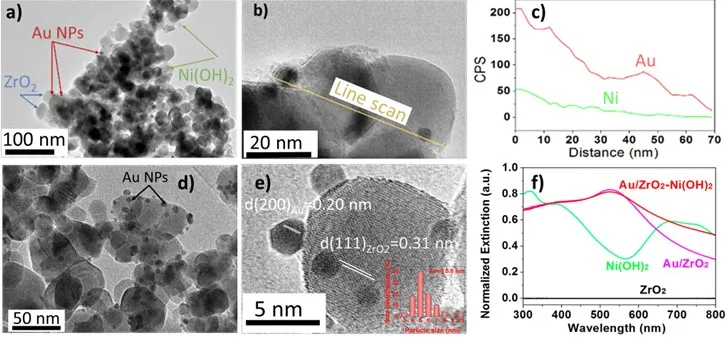
a contains a transmission electron microscopy (TEM) image of a 1 wt% Au/ZrO
2–Ni(OH)
2 sample recovered after the reaction. It can be seen that the Ni(OH)
2 formations are attached to Au NPs and to the ZrO
2 support (b). The line scan profile of the sample (c) confirms the co-existence of Au and Ni elements. The Au/ZrO
2 before and after the Ni(OH)
2 assembly exhibited strong absorption in the ultraviolet and visible range of the spectrum (f). The ZrO
2 support has a large band gap of 5.8 eV and does not absorb light with wavelengths above 400 nm [
22]. The initially prepared Ni(OH)
2 solution absorbed light with two broad peaks observed at the 382 and 690 nm, assigned to the 3*A*
2*g*(*F*)→3T
1*g*(*P*) and 3*A*
2*g*(*F*)→3*T*
1*g*(*F*) transitions of Ni (II) [
23,
24,
25]. The Au/ZrO
2–Ni(OH)
2 catalyst absorbed light in the visible region with the characteristic LSPR absorption peak centered at 525 nm due to the LSPR effect of Au NPs (with diameters about 5 nm, d,e) [
26]. More characterization data are given in SI, Table S1 and Figures S1–S4.
. (a) and (b) TEM image of 1 wt% Au/ZrO2–Ni(OH)2 catalyst. (c) EDX spectrum line profile analysis of 1 wt% Au/ZrO2–Ni(OH)2 indicated along the yellow line in b. (d) TEM image of the 1 wt% Au/ZrO2 catalyst. (e) HRTEM of Au NPs (particle size distribution is inserted). (f) UV-vis spectra for 1 wt% Au/ZrO2 and Au/ZrO2–Ni(OH)2 catalysts.
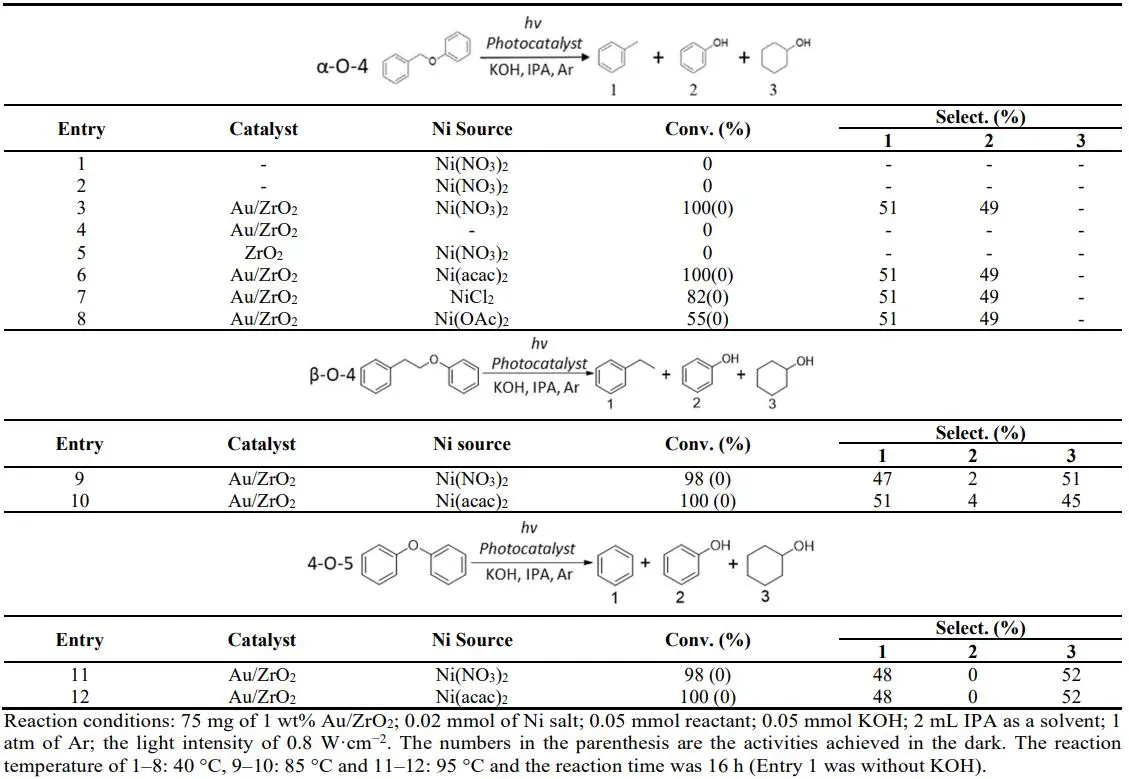
The photocatalytic performances of the catalysts are provided in . Optimal photocatalytic activity toward the lignin model compound α-O-4 (dissociation energy 218 kJ/mol) [
27] cleavage was observed with the structure composed of 1 wt% Au with Ni(OH)
2 on ZrO
2 support, where complete conversion of benzyl phenyl ether and high product selectivity were observed under mild conditions and light irradiation.
. Photocatalytic reaction lignin models catalyst and reaction condition optimization.
Ni(NO
3)
2 dissolved in IPA did not exhibit any activity (Entry 1), and adding 1 equivalent of KOH into the reaction did not lead to reactant conversion (Entry 2). Ni(NO
3)
2 precipitated to Ni(OH)
2 in the solution immediately (Figure S5). When 1 wt% Au/ZrO
2 was added to the reaction, 100% reactant conversion was achieved, producing the products toluene and phenol at a 1:1 ratio at only 40 °C in 16 h (Entry 3). Au/ZrO
2 alone did not lead to a reaction under the same reaction conditions (Entry 4), confirming the synergetic effect of Au/ZrO
2 with Ni(OH)
2 and light irradiation for catalyzing the reaction. Notably, this synergetic effect is induced by light irradiation, as can be seen by comparing to all the control reactions conducted at the same temperature but without light, which exhibited no reaction activity. ZrO
2 and Ni(OH)
2 on their own (Entry 5) showed no activity either.
We note that when different Ni salts, Ni(acac)
2, NiCl
2 and Ni(OAc)
2, were used, there was a photo-induced catalytic activity towards α-O-4 cleavage in the presence of Au/ZrO
2 (Entry 6–8). The difference in reaction activity within different Ni salts may be due to different nanostructures of Ni(OH)
2 formed from different Ni salts [
28].
In the photocatalytic conversion of the model molecule with a β-O-4 linkage, the largest fraction of ether bonds in lignin has a high C–O bond dissociation energy of 289 kJ/mol, and the model molecule with a 4-O-5 linkage has a C–O bond dissociation energy of 314 kJ/mol [
27], over Au/ZrO
2–Ni(OH)
2 are presented in Entry 9–12 of . The result is promising, with 100 % photo-induced conversion achieved at elevated reaction temperatures. The higher temperature is required to achieve the same outcome due to the higher bond energies of the C–O bond of β-O-4 and 4-O-5 linkages. Plasmonic NPs can couple absorbed light and thermal stimuli to generate hot electrons with high energies [
18,
19,
20,
21]. The reaction could proceed when the sum of light and thermal energy absorbed by the catalyst was sufficient to overcome the reaction activation energy barrier. The product selectivity observed in reactions with those model reactant compounds containing β-O-4 and 4-O-5 cleavage was towards cyclohexanol rather than phenol, as observed with α-O-4 cleavage under higher temperatures and intense light conditions. This indicated that after the C–O bond was cleaved at the catalyst surface, phenol was further reduced to cyclohexanol. The cleaving of the 4-O-5 linkage represents an exigent challenge; the catalysts we reported upon previously could not achieve it [
18]. There was no conversion for both lignin model molecules in dark reactions, which confirms the mechanism of action involved in photo-induced catalytic reactions.
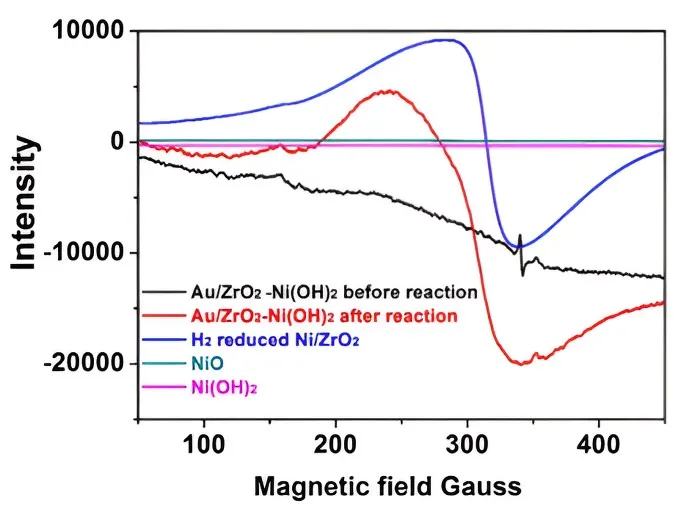
We observed that the catalyst color changed from light purple to dark grey before and after the reaction (Figure S5) and performed EPR analysis on the catalyst samples before and after the reaction, which was also compared with a sample of H
2-reduced Ni NPs loaded on ZrO
2 (). The characteristic peak of paramagnetic Ni
0 was observed, similar to when H
2-reduced Ni NPs were loaded on ZrO
2. This indicates that Ni
0 was the intermediate active site formed during the Ni
2+-Ni
0-Ni
2+ cycles. This is consistent with our previous study [
18]. Light irradiation is known to generate energetic hot electrons in Au NPs and these photogenerated charges transfer to nearby Ni
2+ ions and reduce them to Ni
0 atoms. The system produced from Ni(acac)
2 exhibited the best catalytic performance (Entries 10 and 12). Acetylacetonate molecules which have a conjugated π orbital system, can act as electronic bridges to transfer hot electrons from illuminated Au NPs to Ni(II) and form the catalytic active Ni
0 sites [
18].
. EPR spectra of catalysts and reference materials before and after the reaction.
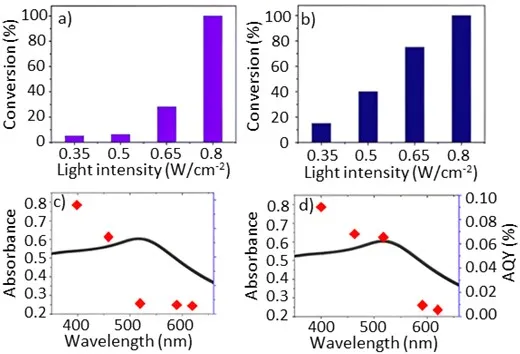
To better understand how light irradiation influenced photocatalytic performance, a study of the reaction rate dependence on light intensity and wavelength at two different reaction temperatures was investigated. a,b shows that the reaction rate considerably increased when the light intensity was increased. At 40 °C, only with high-intensity light irradiation above 0.65 W/cm
2 could the reaction progress with significant reactant conversion. 100% conversion was observed when the light intensity was further increased to 0.8 W/cm
2 at 40 °C.
Increasing the light intensity during the reaction is expected to produce more hot electrons and thus more Ni
0 sites that induce the reductive cleavage [
18,
26], accelerating the reaction. A study was made of the effect of light intensity, performed at two different reaction temperatures, 40 °C (a) and 50 °C (b), using the broad-spectrum CW halogen light source which irradiated the samples over a 400–750 nm wavelength range. At 40 °C reaction temperature, the reactant conversion at low light intensity (0.5 W/cm
2) was measured to be only 4% (a). At a temperature of 50 °C, 0.5 W/cm
2 low intensity light induced 39 % conversion of the reactant (b). The reaction temperature clearly had a marked effect on the photocatalytic cleavage reaction. Changes to the reaction temperature simultaneously influence several physical properties of the system, including diffusion, reactant adsorption and product desorption. Higher temperature also populates higher energy vibrational states of the surface-bound reactant, which can release energy into the reaction coordinate [
29].
The linear dependence indicates a hot electron-driven chemical reaction [
30]. In comparison, a nonlinear dependence of the conversion rate on the light intensity is observed at 40 °C (a). This indicates that the reaction rate is also determined by other effects in addition to hot electrons, such as reactant diffusion to the active sites and the product diffusion from the catalyst and their adsorption on and desorption from the catalyst. At 50 °C, the diffusion and desorption are faster than at 40 °C. The reaction rate predominantly depends on the number of hot electrons. A linear dependence on the light intensity is observed (b).
A one-to-one action spectrum mapping was performed between the wavelength-dependent photocatalytic rate and the light extinction spectrum. c,d demonstrates that the irradiation wavelength has a crucial impact on the photocatalytic C–O bond cleavage of α-O-4. We note that the trend of quantum yields does not follow the trend of light absorption at 50 °C. The reaction is only activated at wavelengths shorter than 450 nm. The 395 nm wavelength photons have an energy of ~3.12 eV, which provides sufficient energy to the excited electrons to overcome the energy barrier for the reduction of nearby Ni
2+ in Ni(OH)
2 to Ni
0 [
18]. The population of excited electrons is greatly boosted near the LSPR wavelength range, but the electrons excited by the LSPR absorption have insufficient energy to reduce Ni
2+ ions and initiate the C–O bond cleavage in α-O-4. These indicate there is an energy threshold to drive the photocatalytic reaction [
29]. Hence, at 50 °C or below, the photocatalytic reactions are driven by the hot electrons of the Au NPs, generated by interband electron excitation [
18]. At a higher temperature, the population of excited vibrational states of the reactant molecules is greater. It means a reduced activation energy barrier [
31]. Also, there are more conducting electrons of plasmonic metal NPs at higher energy levels [
19], which can be excited and transferred to Ni
2+ sites by photons of longer wavelengths. The energy threshold decreases as the reaction temperature is raised [
30]. When the reaction temperature increased to 65 °C, the AQY at the wavelength in the LSPR range (525 nm) showed a great increase (d). The light energy in LSPR wavelength can effectively drive the reaction since the LSPR absorption of Au NPs generates a much more hot electrons with energy exceeding the threshold, inducing the reaction. It reveals that moderate heating, which can be achieved by absorption of infrared irradiation of the solar spectrum, will significantly enhance the catalytic performance under irradiation of visible light that accounts for about 45% of the sunlight energy. This knowledge is useful for driving the reaction with sunlight in the future.
. Light intensity dependence on photocatalytic cleavage of α-O-4 lignin model molecule using 1 wt% Au/ZrO2–Ni(OH)2 catalyst at different temperatures at (a) 40 °C for 16 h and (b) 50 °C for 8 h. Action spectra shows that the dependence of the catalytic activity of α-O-4 cleavage on the different wavelength with 0.2 W/cm2 intensity at (c) 50 °C and (d) 65 °C.
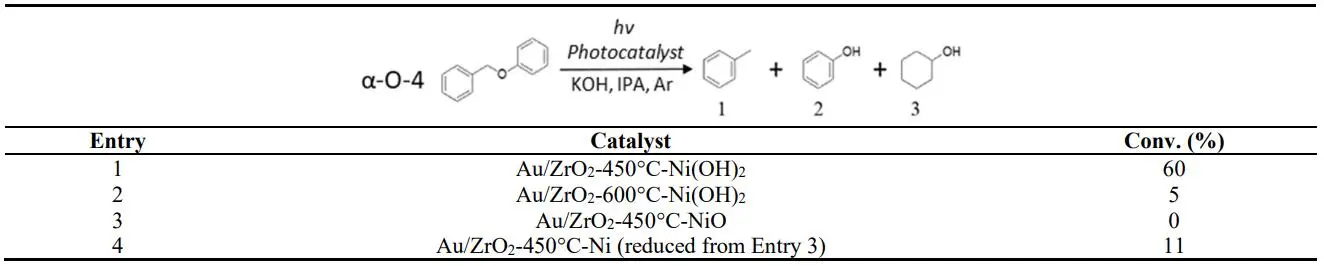
To explore the possible mechanism, 1 wt% Au/ZrO
2–Ni(OH)
2 was calcined at 450 °C in the air for 1 h. In the calcining process, Au NPs aggregated to a larger size at higher temperatures and Ni(OH)
2 decomposed to NiO, as confirmed by the FTIR spectrum in Figure S6 (the obtained catalyst is labeled as 1 wt% Au/ZrO
2-450-NiO). For comparison, 1 wt% Au/ZrO
2 was calcined at 450 °C for 1 h first to achieve the Au NPs of similar sizes. The calcined sample was then used to prepare 1 wt% Au/ZrO
2-450-Ni(OH)
2 catalyst by precipitating Ni(OH)
2 on it with KOH in Ni(NO
3)2/IPA solution. The difference between the two catalysts is that one contains NiO and the other Ni(OH)
2, while Au/ZrO
2 in both catalysts were at the same percentage and morphology. The mean size of Au NPs in the calcined samples increased to 7 nm (Figure S7a) compared to the original 5 nm. The 1 wt% Au/ZrO
2-450-Ni(OH)
2 exhibits 60 % conversion (, Entry 1). In contrast, 1 wt% Au/ZrO
2-450-NiO showed no conversion (, Entry 3). It confirms that Ni(OH)
2 played an essential role in catalyzing the reaction. We reduced 1 wt% Au/ZrO
2-450-NiO by H
2 at 550 °C for 30 mins to reduce a part of NiO to Ni
0 sites. FTIR results in Figure S8 supported this reduction. The reduced sample showed 11 % conversion without base added (, Entry 4). These results corroborate that Ni
0 is critical for the reaction. Ni
2+ ions in the Ni(OH)
2 can be readily reduced to Ni
0 atoms when receiving the hot electrons from Au NPs. ZrO
2 support is inert (with a bandgap of 5.8 eV) [
22] and cannot absorb visible light. In contrast, it is difficult to reduce NiO by the hot electrons generated by visible light irradiation on Au NPs to a large number of the active catalytic Ni
0 sites of the reaction.
1 wt% Au/ZrO
2-600-Ni(OH)
2 sample was also prepared by increasing calcination temperature to 600 °C for 4 h. The mean size of Au NPs is 10 nm (Figure S7b), and the sample exhibited only 5 % conversion (, Entry 2). Large Au size is not beneficial for the reaction as the Au surface area and the interface between Au NPs and Ni(OH)
2 decreases.
The effect of Au NP content in a photocatalyst on photocatalytic performance was investigated. We used different amounts of catalysts with different Au loadings to maintain the total Au content in the catalytic systems the same at 75 mg and used identical reaction conditions otherwise.
. Photocatalytic performance of C–O bond cleavage of the α-O-4 model with Au NPs on different supports.
Figure S9a shows that the higher the Au NP density (higher Au content) on the ZrO
2 support, the lower the conversion rate of the reaction. UV-Vis spectra in Figure S3 show that the higher Au content leads to greater light absorption. However, the catalytic activity of the sample with the higher Au content decreased. For example, 5 wt% Au/ZrO
2 gives 52% conversion, whereas 10 wt% Au/ZrO
2 only gives 3 % conversion. TEM results in Figure S2 indicate that the higher Au loading results in an increase in particle size while the specific surface area of Au NPs decreases. There are fewer Au NP hot spots, where the density of hot electrons and EM field intensity are likely to be high with larger AuNP particle size samples. The contact between Ni(OH)
2 and Au NP surface at hot spots decreases, given the same total Au amount in the catalyst. This is likely to reduce the transfer of the hot electrons from Au NPs to Ni(OH)
2, resulting in fewer Ni
0 atoms and weaker catalytic performance.
When maintaining the Au content constant and reducing the Ni
2+ cation amount from 0.02 mmol to 1.25 μmol (Figure S9b), we found that the catalytic conversion drops significantly to 10 %. Since reduced Ni
0 sourced from Ni(OH)
2 is the catalytic site for the cleavage reaction, less Ni
2+ in the reaction medium results in insufficient catalytic sites to drive the catalytic reaction. Au/ZrO
2 and Ni(OH)
2 work synergistically to catalyze the reaction, and an optimal ratio between the two is required. Figure S9b also shows when the amount of 1 wt% Au/ZrO
2 increases while the amount Ni
2+ in the reaction system is maintained constant, the conversion rate increases; 75 mg Au/ZrO
2 with 0.01 mmol of Ni
2+ cations achieves the best result at 100 % conversion. Less amount of Au/ZrO
2 means fewer hot electrons generated under visible light irradiation, which gives lower catalytic activity.
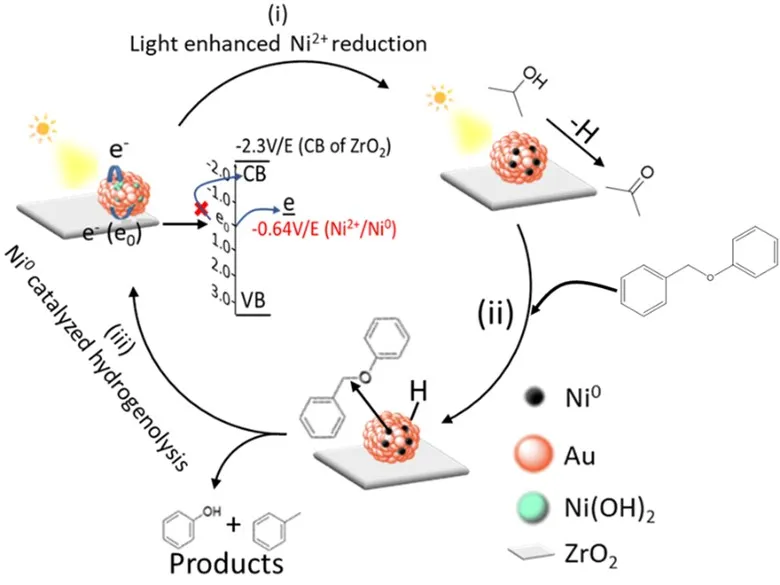
Based on the above results, we proposed a tentative reaction pathway for the lignin compound C–O bond cleavage catalyzed by Au/ZrO
2–Ni(OH)
2 under visible light irradiation as depicted in . The solvent and reducing agent/solvent IPA absorbed on Au NPs is oxidized to acetone over Au NPs, releasing active H species and electrons that quench with the holes generated by light irradiation on Au NPs [
20]. The basic medium maintained by adding KOH provides the conditions suited to the abstraction of H atoms from isopropanol. Light excites hot electrons in the Au NPs, which transfer to Ni(OH)
2 and reduce Ni
2+ to Ni
0. Ni
0 can insert into the C–O bond through oxidative addition and cleave the bond via a hydrogenolysis process but be oxidized back to Ni
2+ [
32]. After the reductive elimination, the Ni
2+ site is reduced to Ni
0 in the subsequent catalytic cycle. A possible reason for the excellent performance of these catalysis systems for the 4-O-5 linkage cleavage is that the active catalytic Ni
0 sites in these systems have better accessibility to the ether C–O bond of the reactant, compared with those immobilized on alumina fibers.
The possibility of catalyst recycling is a key criterion for heterogeneous catalysts. Experiments were carried out to demonstrate the recyclability of the Au/ZrO
2–Ni(OH)
2 catalyst. The catalyst from the first run was washed with IPA three times, dried in the vacuum oven, and reused without adding further KOH. Au/ZrO
2–Ni(OH)
2 exhibits good reusability, as shown in Figure S10. The catalyst was reusable for several cycles.
. A proposed mechanism for photocatalytic lignin model cleavage with Au/ZrO2–Ni(OH)2 under visible light irradiation.
The following supporting information can be found at: https://www.sciepublish.com/index/journals/article/prp/28.html/id/16.
Conceptualization, H.Z.; Methodology, Y.J. and X.W.; Validation, X.W.; Formal Analysis, Y.J., X.W., Y.H.; Investigation, X.W. and Y.J.; Resources, H.Z. and E. R.W.; Writing—Original Draft Preparation, Y.J.; Writing—Review & Editing, Y.J., X.W., E.R.W. and H.Z; Supervision, E.R.W and H.Z.; Project Administration, H.Z.; Funding Acquisition, H.Z and E.R.W.
Not applicable.
Not applicable.
This research was funded by the Australian Research Council for Discovery Projects DP20010265 and DP210103357.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.