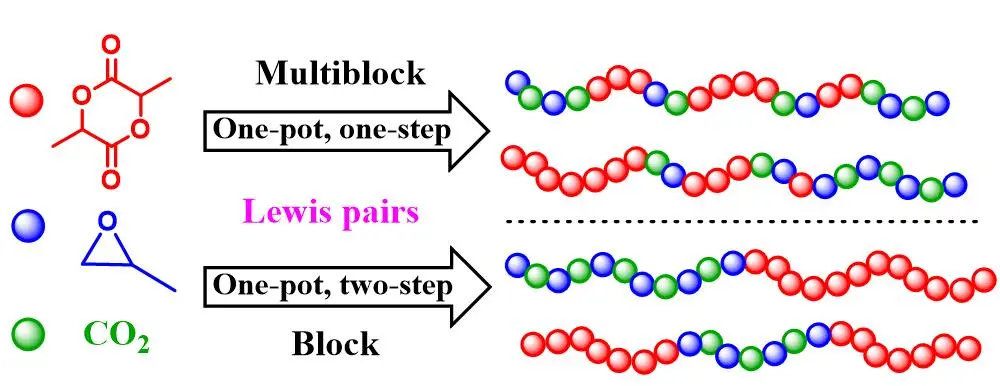
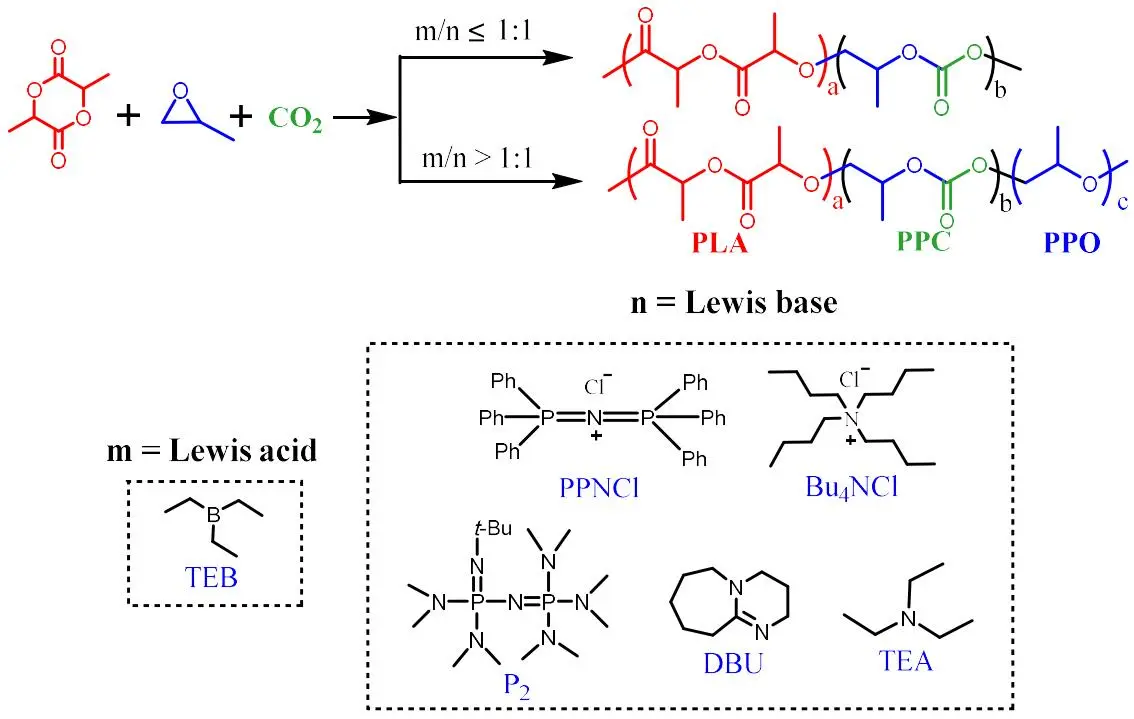
1. Introduction
Due to the severe ecological and environmental issues caused by improperly discarded non-degradable plastics, there is an urgent demand to develop eco-friendly biodegradable polymer materials [
1,
2,
3]. Aliphatic polyesters and polycarbonates are two classes of biodegradable polymers among which polylactide (PLA) and poly(propylene carbonate) (PPC) are two typical polymers extensively studied in academia and industrialized as biodegradable resins [
4,
5]. PLA, as a potential non-petroleum-based substitution of fossil-based polyesters, suffers from brittleness upon physical aging and thus its prospective applications are inevitably restricted [
6]. For overcoming its drawbacks, numerous modification strategies [
7] such as blending [
8] and copolymerization [
9] have been exploited. As for PPC, a high-value-added CO
2-based material, has great potential in practical applications [
5], but its low glass transition temperature (*T*
g) and amorphous nature cause the problem of cold flow [
10]. To help to resolve the dilemmas, it is feasible to combine the crystallization property of PLA and the toughness of PPC by the terpolymerization of propylene (PO), lactide (LA) and CO
2 and the construction of PPC-PLA block copolymer.
The terpolymerization of PO/LA/CO
2 in one-step strategy usually produces random, gradient or multiblock terpolymers. PO/LA/CO
2 terpolymer (PPCLA) has stronger degradability due to the existence of the lactide ester unit [
11]. Our group synthesized PPCLA with long LA-rich sequence catalyzed by zinc adipate and revealed that 12% LA molar content in the polymer was enough to endow PPCLA with crystallinity [
12]. With the increasing amount of LA content, the *T*
g and thermal stability of PPCLAs are all higher than that of PPC [
13]. Except for heterogeneous metal catalyst, a binary [
14] or ternary [
15] homogeneous catalytic system with cocatalyst and conjugated tri-nuclear salen-Co complexes without cocatalyst [
16] are applied in this one-pot terpolymerization. But inevitable transesterification was reported in these catalyst systems. To our knowledge, the main chain structures of the terpolymer via one-step strategy are not the regular AB or ABA types. So the two-step procedure is necessary when synthesizing di- or tri-block copolymer. Darensbourg et al. demonstrated that a tandem catalytic approach was feasible for the synthesis of model A-B CO
2 diblock copolymer [
17]. The key to this strategy is choosing water as a chain-terminating reagent after polycarbonate synthesis to generate monohydroxy macroinitiators in situ, which allows direct organocatalytic LA ring-opening polymerization [
18]. In addition, they synthesized triblock copolymer poly(lactide-*b*-propylene carbonate-*b*-lactide) (PLA-PPC-PLA) where water acts as a chain-transfer reagent along with the PO/CO
2 copolymerization process to produce polyol [
19,
20]. What’s more, gas-controlled polymerization was intelligently developed to switch whether the self-polymerization of LA proceeded or not by reversible absorption of CO
2 into organic amidine [
21]. Considering the catalytic systems mentioned above, the synthesis of PPCLA and block polymers of PO/LA/CO
2 are based on metal catalytic processes, although that of PLA block is based on organic one. So it’s interesting to replace metal catalysts with metal-free one to synthesize this polymer, avoiding the undesired metal residual.
In 2016, a significant breakthrough made by Feng et al. was to employ triethyl borane (TEB) and quaternary ammonium salt as a metal-free Lewis pair catalyst in the synthesis of CO
2-based polycarbonate [
22]. After this pioneer work, TEB/organic base pairs make great success in the synthesis of polycarbonates [
23,
24,
25], polyethers [
26,
27,
28,
29], polyesters [
30,
31,
32,
33,
34], poly(ether-ester)s [
35,
36,
37,
38], poly(ester-carbonate)s [
39,
40,
41,
42] and others [
43]. Inspired by these works, TEB/organic base pairs are selected to catalyze the terpolymerization of PO/LA/CO
2 for the first time. PO/LA/CO
2 terpolymerization consists of two kinds of reaction types: one is the ring-opening polymerization (ROP) of LA, and the other one is the ring-opening copolymerization (ROCOP) between CO
2 and PO. Herein, TEB/bis(triphenylphosphine)iminium chloride (PPNCl) pair was chosen as the model catalyst to optimize reaction conditions. Although this pair prevails in polycarbonate synthesis, it is a new catalytic system for the ROP of LA. Hence, we explored how catalyst worked in PLA synthesis as the first step, and then, one-pot one-step and one-pot two-step strategies were adopted in the terpolymerization. In the one-step strategy, even when the molar ratio of PPNCl to TEB was greater than 1, the terpolymer PPCLA was successfully synthesized. When employing the two-step tandem strategy, the well-defined PPC-PLA and PLA-PPC-PLA block copolymers were simply constructed with a main difference from the reported ones, because no additional organic catalyst was needed in the subsequent ROP of LA.
2. Materials and Methods
*2.1. Materials and Instruments*
All chemicals were used as received unless otherwise mentioned. Propylene oxide (PO, 99%, Energy, Shanghai, China) was dried on 4 Å molecular sieve for more than 24 h before use. Bis(triphenylphosphine)iminium chloride (PPNCl, Alfa, Waltham, USA, 97%), tetrabutylammonium chloride (NBu4Cl, Alfa, 98%), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, Aladdin, Shanghai, China, 98%), triethylamine (TEA, Aladdin, Shanghai, China, 99%), phosphazene base P2-*t*-Bu (P2, Sigma-Aldrich, City of Saint Louis, USA, 2 M in THF), triethyl borane (TEB, Energy, 1 M in THF), 1,4-benzenedimethanol (BDM, TCI, 98%), benzyl alcohol (BnOH, TCI, Tokyo, Japan, 98%), ʟ-lactide (LA, Purac, The Netherlands, >99.5%) and high purity CO2 (Guangqi Gas Co. Ltd., Guangzhou, China, >99.9999%) were used as received.
1H NMR and diffusion-ordered NMR spectroscopy (DOSY) spectra of PLA, PPCLA and PPC-PLA in deuterated chloroform (CDCl3) were recorded on a Bruker DRX-500 MHz NMR spectrometer with tetramethylsilane (TMS) as an internal reference.
Matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI-TOF MS) experiments were carried out on Bruker Ultrafle Xtreme. 1:1 volume ratio of polymer sample (1 mg/mL in THF) and trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) matrix (10 mg/mL in THF) were mixed, and then 1 μL mixed solution was spotted onto the sample plate for the test.
Molecular weights were tested by a waters gel permeation chromatography (GPC) system equipped with Shodex K series and Waters 2414 using chloroform as eluent (1 mL/min at 40 °C) with narrow Mw polystyrene (PS) as standards.
Thermogravimetric analysis (TG) was performed on a PerkinElmer Pyris Diamond TG/DTA analyzer between 50 °C and 500 °C with a heating rate of 10 °C/min in nitrogen (150 mL/min). Each sample for the test was about 10 mg.
Differential scanning analysis (DSC) measurements were carried out on a DSC Model 204 (Netzsch) under nitrogen flow in the temperature range of −10–110 °C. The samples (around 8 mg) were firstly heated at a heating rate of 10 °C/min and then cooled at a cooling rate of 10 °C/min by liquid nitrogen, followed by second heating. The glass transition temperature (Tg) was measured from the second heating cycle.
*2.2. Typical Procedure for ROP of LA*
Take Entry 9 in as an example. In a glovebox, PPNCl (57.6 mg, 0.1 mmol), LA (1.440 g, 10 mmol) and PO (2.904 g, 50 mmol) were measured into a 20 mL glass bottle with a stir bar. TEB (1 mmol/mL in THF, 100 µL, 0.1 mmol) was then carefully added to the glass bottle via syringe. Sealed the glass bottle and took it out of the glove box followed by stirring at 25 °C for 30 min. A small aliquot of the polymerization mixture was taken out for 1H NMR test. The reaction was quenched by HCl in ethanol (1 mol/L) and then dissolved in 2 mL CH2Cl2. Finally, the polymer mixture was precipitated in an 8-fold excess of ethanol, which was dried in vacuum at 60 °C until a constant weight.
*2.3. Typical Procedure for the One-Pot One-Step Terpolymerization of PO/LA/CO2*
First, the 50 mL autoclave was dried in an oven at 120 °C overnight, then immediately placed into the glovebox. When it cooled to room temperature, the copolymerization was described below. Taking Entry 1 in as an example, PPNCl (57.6 mg, 0.1 mmol), LA (1.440 g, 10 mmol) and PO (2.904 g, 50 mmol) were measured into the reactor and then TEB (200 μL, 0.2 mmol) was carefully added. The reactor was sealed and taken out from the glove box and charged with CO2 under a pressure of 1 MPa. The copolymerization was carried out at 60 °C for 10 h in the oil bath and the CO2 pressure was kept at consistent 1 MPa. After 10 h, cooled the reactor in running water, released the CO2, and took out a drop of mixture for 1H NMR test. Added 3 mL CH2Cl2 to dissolve the crude product and quenched the reaction with 0.25 mL HCl in ethanol (1 mol/L). The polymer solution was precipitated in 8-fold excess of ethanol, and the product was dried in vacuum at 60 °C for 24 h until no weight change.
*2.4. Typical Procedure for the One-Pot Two-Step Strategy to Synthesize Block Copolymer*
Taking Entry 4 in as an example, PPNCl (28.8 mg, 0.05 mmol), PO (2.904 g, 50 mmol) and TEB (100 μL, 0.1 mmol) were added into the 50 mL autoclave in a glovebox. The reactor was sealed and taken out from the glove box and charged with CO2 under a pressure of 1 MPa. The copolymerization was carried out at 60 °C for 2 h in the oil bath and kept at the CO2 pressure of 1 MPa. And then cool the reactor in ice water to the room temperature. Took the autoclave into the glovebox to release the unreacted CO2 slowly, and opened it to take some mixture for 1H NMR and GPC tests. After LA (720 mg, 5 mmol) was added, the reactor was sealed and taken out of the glovebox for another 1.5 h reaction in 60 °C oil bath. When the reaction came to an end, immersed the reactor in ice water bath until it was cool down, and a small aliquot of the polymerization mixture was taken out for 1H NMR test. The reaction was quenched with HCl in ethanol (1mol/L). The crude product was dissolved with 3 mL CH2Cl2 and then precipitated in 8-fold excess of ethanol, collected, and dried in vacuum at 60 °C until a constant weight.
3. Results and Discussion
*3.1. Synthesis of PLA*
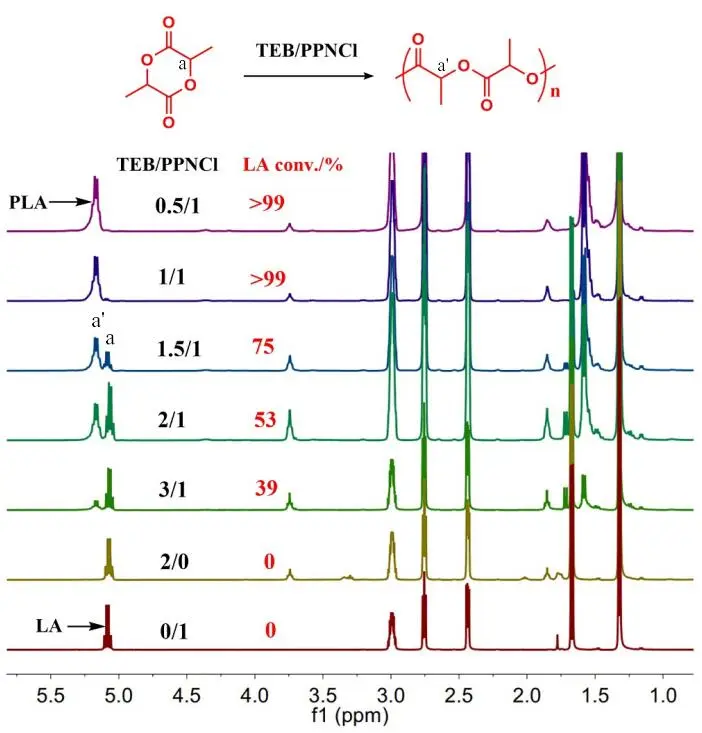
Prior to delineating the terpolymerization of PO/LA/CO
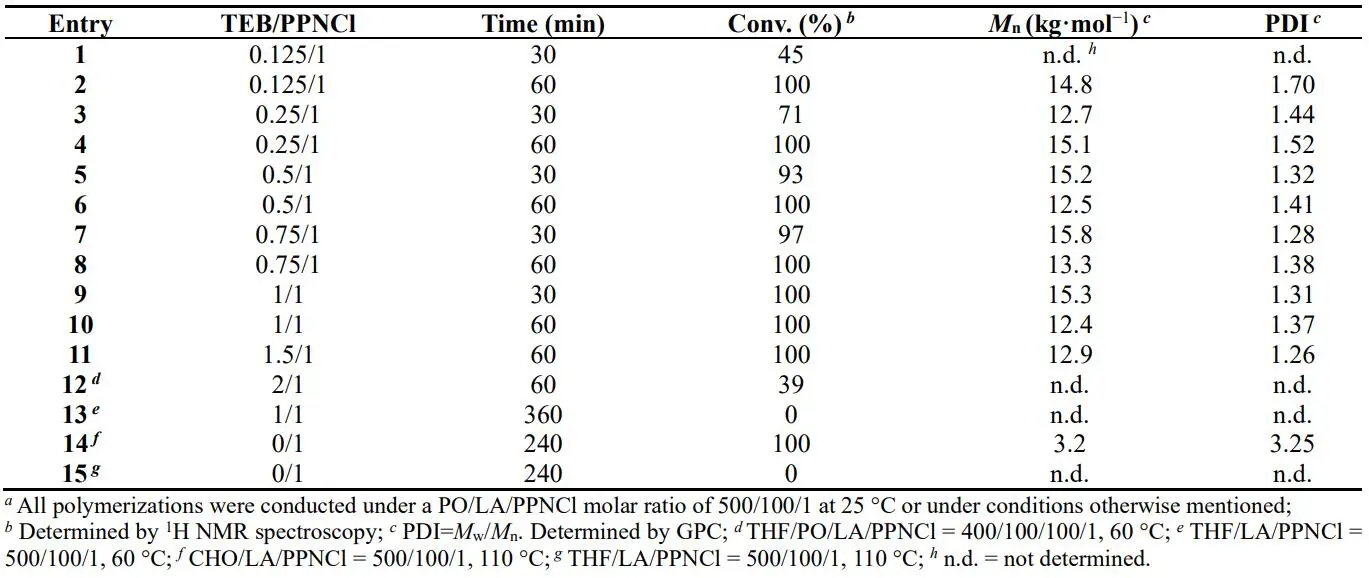
2, the ROP of LA catalyzed by TEB/PPNCl Lewis pair was adequately studied. A series of simple experiments were designed to screen the suitable TEB/PPNCl ratio for optimal reaction activity as illustrated in . As expected, no polymer was formed if there was sole TEB or PPNCl utilized as catalyst. Varying the TEB/PPNCl ratios from 0.5/1 to 3/1, the existence of TEB greatly hindered the catalytic efficiency in the ROP of LA, which is similar to the results catalyzed by TEB/1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) pair [
31]. But within 10 min, there seemed no differences in the reaction activity between 0.5/1 and 1/1 TEB/PPNCl ratios. This phenomenon pushes us to explore what would happen if the TEB/PPNCl ratio is less than 1. So we conducted the experiments at 25 °C for a better comparison in the reaction rate. Fixing the molar ratio of PO/LA/PPNCl = 500/100/1 and increasing TEB from 0.25 to 1, LA displayed an improved conversion rate in 30 min (, entries 1, 3, 5, 7 and 9), suggesting TEB/PPNCl = 1 is the optimal ratio. If replaced PO with tetrahydrofuran (THF) as the solvent to dissolve LA, no polymer was detected in
1H NMR even reacting at 60 °C for 6 h with a TEB/PPNCl ratio of 1/1 (, Entry 13). But when mixing some PO into THF as solvent (, Entry 12), PLA was synthesized at a lower reaction rate in comparison with only PO as solvent (). Besides PO, cyclohexene oxide (CHO) was tested as a good solvent in this catalytic system. Considering the difference between THF and two epoxides, THF usually serves as the solvent in the ROCOP of PO/CO
2 and CHO/CO
2 catalyzed by TEB/PPNCl, where Cl
− attacks PO and CHO to ring open them as the initiation step, meaning that THF is inactive in this catalyst system.
To the best of our knowledge, there is no report about chlorine salt catalysts in the ROP of LA due to the weak nucleophilicity of chloride ion for ring opening LA ring. Inspired by the copolymerization of epoxide and anhydrides catalyzed by PPNCl, where chloride ion was able to ring open CHO as the initial step in polyester synthesis at 120 °C [
44], we tried to synthesize PLA mediated by PPNCl using CHO as solvent. The experiment result demonstrated that PPNCl was a feasible base catalyst for PLA synthesis initiated by the formation of oxygen ion active species (, Entry 14). However, severe transesterification leads to the low molecular weight (*M*
w) and the broad polymer dispersion index (PDI). When replaced CHO with THF as solvent under the same reaction condition (, Entry 15), no PLA signal was detected by
1H NMR. So the real initiator for PLA ROP is the oxygen ion active species formed by the ring opening of CHO and Cl
−.
To further discover the differences between TEB/PPNCl = 0.5 and TEB/PPNCl = 2 on the polymer synthesis, the reaction procedures were monitored by
1H NMR technique. Comparing the reaction time in Figure S1 and Figure S2, longer time should be spent on the majority of LA conversion no matter at 25 °C or 60 °C when TEB/PPNCl = 2. What’s more, the splitting of PLA peak in TEB/PPNCl = 0.5 becomes unclear with time at the reaction temperature of 25 °C but this phenomenon doesn’t occur in TEB/PPNCl = 2, suggesting that the excess TEB helps to suppress the transesterification with the sacrifice of reaction rate. Besides the mentioned above, no poly(propylene oxide) (PPO) signal was detected when TEB/PPNCl = 0.5 even if extending the reaction time to 6 h at 25 °C, however, the result of TEB/PPNCl = 2 was the opposite.
. Results of ROP of LA catalyzed by different TEB/PPNCl ratios *a*.
. Evolution of 1H NMR spectra for the copolymerization of PO/LA/PPNCl (500/100/1) catalyzed by different ratios of the TEB/PPNCl pair at 60 °C for 10 min.
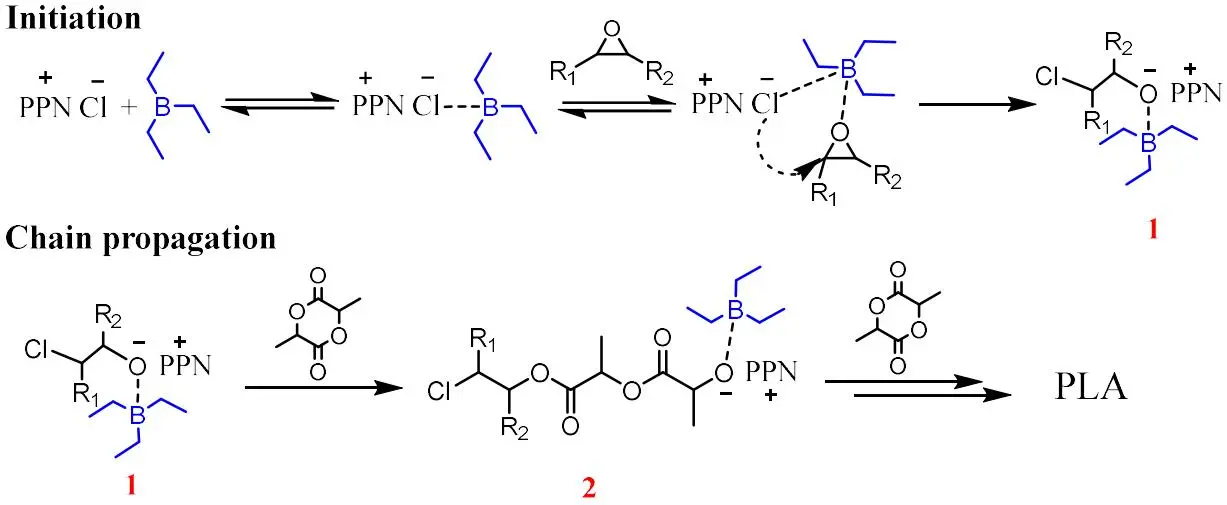
Based on these observations, the proposed mechanism when TEB/PPNCl = 1 in the ROP of LA is shown in . In the initiation step, the binding of PPNCl to TEB facilitates the ring opening of PO first to form PPN(OCH(CH
3)CH
2)nCl (species 1) stabilized by TEB; then LA inserts into species 1 continuously to form PLA chain (species 2) terminated with −(OCH(CH
3)CH
2)nCl as the chain propagation step. The overwhelming value of n in PPN(OCH(CH
3)CH
2)nCl is calculated to be 1 by matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI-TOF MS) studies (Figure S3). It’s hard to ignore that the reaction rate decreases accompanying with the alleviative transesterification when increasing the TEB usage from TEB/PPNCl = 1. Based on that fact, a reasonable explanation can be drawn as that, the excess TEB combines with PO to deliver active species TEB-PO and then TEB-PO competes with LA in the insertion into the active chain terminal, leading to the formation of PPO (Scheme S1, path 3) and the lower reaction rate in path 1. As for the undesired transesterification (Scheme S1, path 2), it’s weakened by the growing TEB content with the ability to stabilize the anion terminal but the formation of PPO is inevitable. If TEB/PPNCl < 1, TEB is not enough to stabilize the oxygen anion terminal and helps to increase the degree of transesterification, so the conversion of LA decreases. In a word, the best reactivity of the ROP of LA is obtained with a TEB/PPNCl ratio of 1/1.
. Proposed mechanism of PLA synthesis catalyzed by TEB/PPNCl (1/1) pair using PO as solvent.
*3.2. Synthesis of PPCLA in One-Pot One-Step Methodology*
According to the literatures, PPNCl, tetrabutyl ammonium chloride (Bu
4NCl), phosphazene base (P
2) and trimethylamine (TEA) have been used in the synthesis of PPC as the base component of the TEB-based binary catalyst except for DBU [
22,
23]. It is well-known that DBU shows outstanding catalytic activity in PLA synthesis, and our preliminary experimental result proves that TEB/DBU pair catalyst is effective in PPC synthesis. Herein, TEB/PPNCl pair was preferably selected to optimize the reaction temperature and the ratio of TEB/base. As excepted, high temperature contributes to the backbiting of PPC chain leading to the poor selectivity (Table S1). So setting the reaction temperature at 60 °C, different ratios of TEB/PPNCl were tested to screen the proper ratio. According to Table S2 entries 1-6, increasing TEB content favors the formation of PPC and the best selectivity is found in the TEB/PPNCl = 2/1, which is in accord with the ratio revealed by Feng’s group [
45]. One TEB stabilizes the reactive end and the other TEB activates PO during the polymerization. When TEB/PPNCl > 2/1, it can’t help to increase the selectivity or molecular weight. Considering the cost of TEB, TEB is added as little as possible, so TEB/PPNCl = 2/1 is the best ratio in PO/LA/CO
2 terpolymerization with a balance of the reactivity and the cost. This is quite different from the ratio of TEB/PPNCl = 1/1 in PLA synthesis, because unlike PO should be activated in the polymerization, monomer LA reacts smoothly without the aid of TEB. Moreover, PPO is absent when TEB/PPNCl ratio is below 1. However, no polymer except for propylene carbonate was synthesized with the absence of LA at TEB/PPNCl = 0.5, meaning that this catalyst ratio is unable to catalyze the ROCOP of PO and CO
2 (Table S2, Entry 8†). But interestingly, the terpolymerization of PO/LA/CO
2 can proceed smoothly indicating the existence of PO/CO
2 ROCOP although the PPC content is relatively low (Table S2, Entry 1). In addition, an ingenious experiment was designed to declare that the PLA helps the ROCOP of PO/CO
2: synthesized PLA first and then purged CO
2 to synthesize PPC successfully under TEB/PPNCl = 0.5 (Table S2, Entry 7). So the chain propagation process at TEB/PPNCl = 0.5/1 was further proposed as Scheme S2. The polymer chains with a LA end can’t continue to ring-open PO because no ether linkages were observed in the
1H NMR spectrum of polymer (Figure S4, Entry 1). Nevertheless, it can ring-open another LA monomer or be inserted with CO
2 step to form the LA-CO
2 linkage [
15]. When CO
2 takes up the polymer end, only PO can be connected. If a polymer is terminated with a PO end, it will ring-open LA to produce a PO-LA linkage or continue the short PO/CO
2 ROCOP process without the ability of ring-opening PO to form ether linkage. The ROP of LA at TEB/PPNCl = 0.5/1 accompanies with severe transesterification especially when LA monomer is consumed up. The persistent transesterification will produce new LA-end-capped polymer chains, which is conducive to insert CO
2 immediately followed by PO insertion to construct PPC segments. Presumably, this is the reason that no PPC was synthesized in PO/CO
2 copolymerization at TEB/PPNCl = 0.5/1 but PPC was obtained in the presence of LA or PLA.
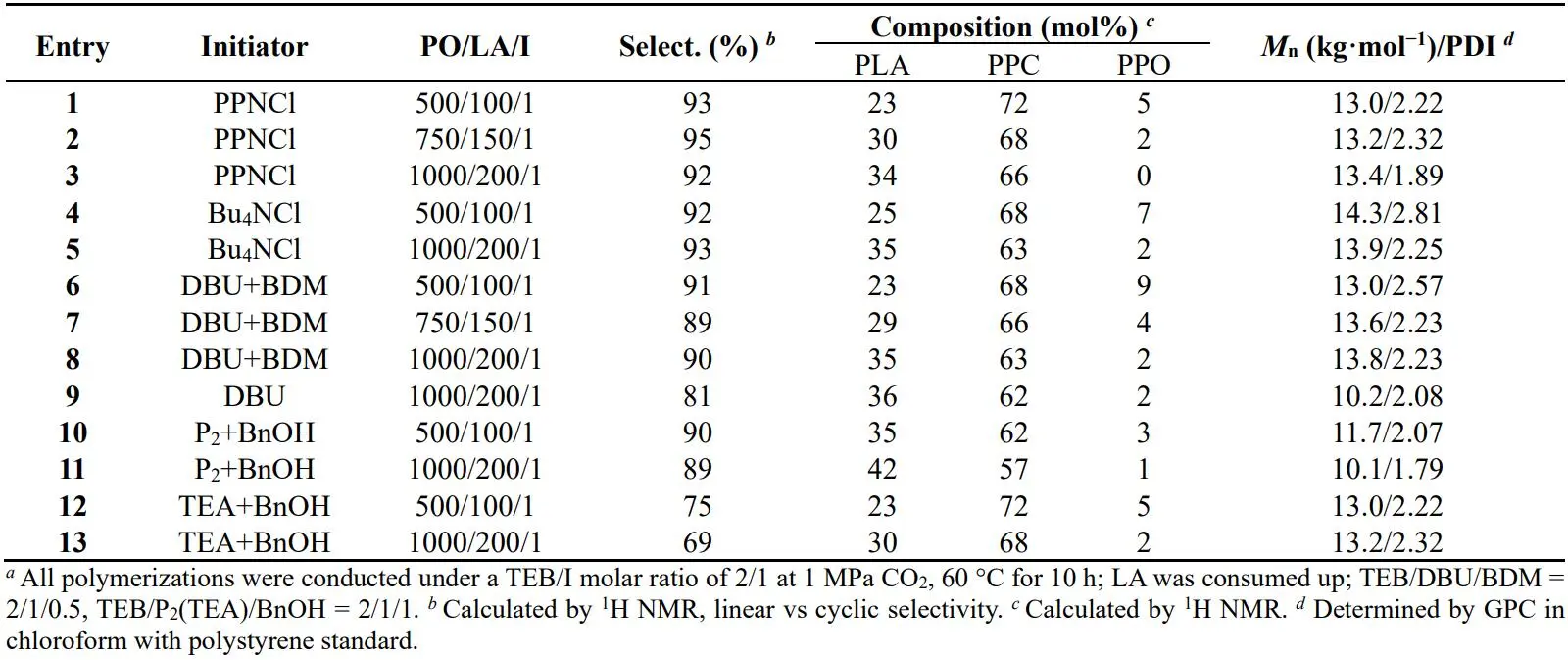
In PPCLA synthesis, five different kinds of bases were used in the terpolymerization (). Quaternary ammonium salt bases display better selectivity as discovered in . It can be obviously observed that all products have low MWs and broad PDIs even when increasing the feeding ratio of monomer to the initiator. No significant change in MW can be observed (, entries 1–3), suggesting the severe transesterification in the terpolymerization of PO/LA/CO
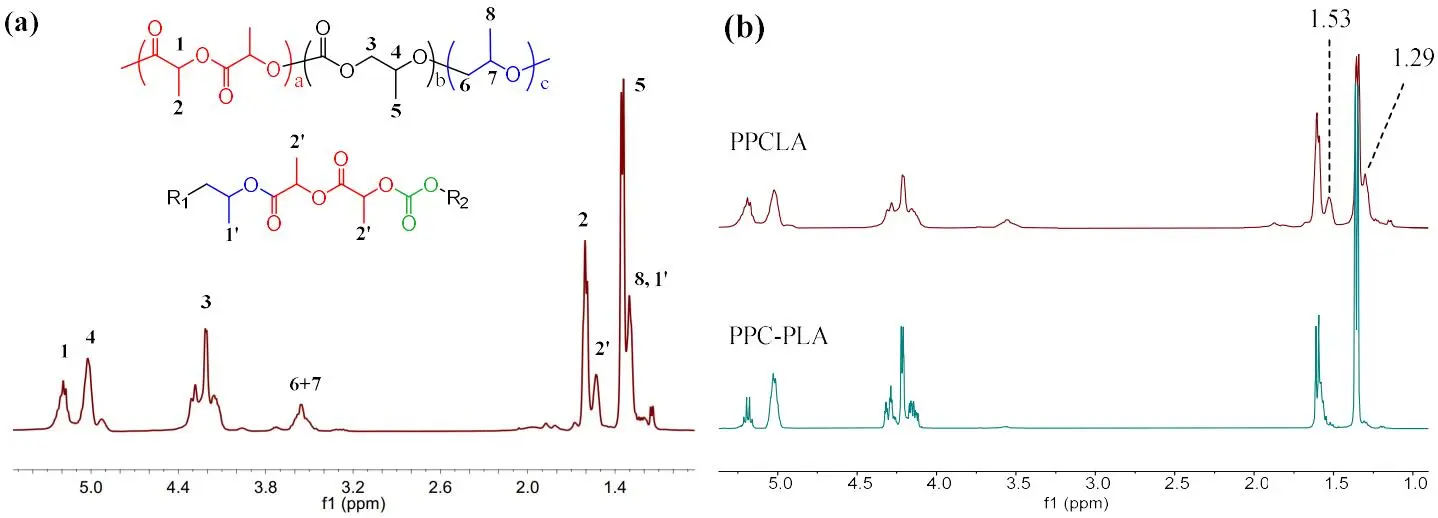
2. The structure of PPCLA was characterized by
1H NMR technic (a), which consists of PLA, PPC and PPO segments. The peaks 1’ (1.29 ppm) and 2’ (1.53 ppm) belong to the CH
3 of PPC units and CH
3 of PLA units respectively generated from PPC-PLA linkages [
16], further proved by no peaks 1’ and 2’ in the
1H NMR spectrum of PPC-PLA block copolymer (b). So the resultant PPCLAs are multiple block copolymers due to the obvious signals of PPC-PLA linkages. To investigate the reaction process in PPCLA synthesis, TEB/PPNCl and TEB/DBU pairs were chosen as modal pairs. The results show that the ROP of LA has a faster reaction rate than the ROCOP of PO/CO
2 and partial PPC was generated before LA was consumed up (Tables S3 and S4, Figures S5 and S6. To unambiguously verify the polymer structure, diffusion-ordered NMR spectroscopy (DOSY) analysis was further performed. Two kinds of PPCLAs synthesized using different catalysts both share the same diffusion coefficient, indicating the successful synthesis of pure copolymer, while a blend of PPC and PLA displays two diffusion coefficients as expected (Figure S7).
. Synthesis of PPCLA catalyzed by different Lewis pairs.
. Synthesis of PPCLA catalyzed by different Lewis pairs *a*.
. (a) <sup>1</sup>H NMR spectrum of PPCLA (, Entry 6); (b) comparison of 1H NMR spectra of PPCLA multiblock copolymer and PPC-PLA block copolymer.
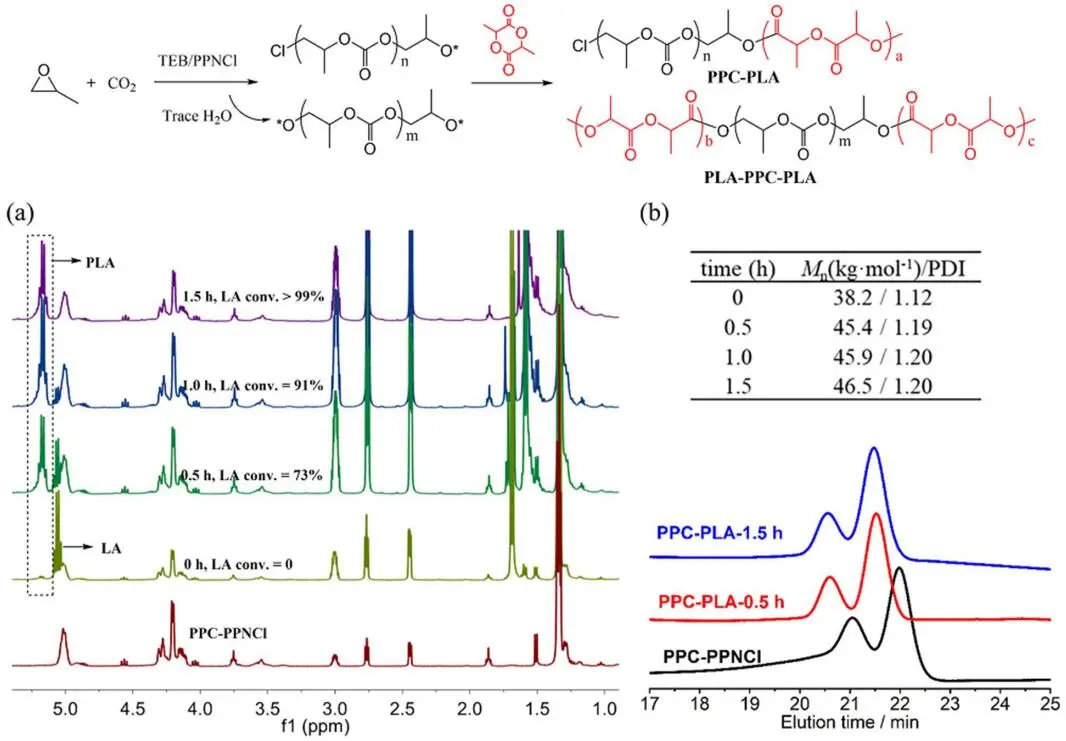
*3.3. Synthesis of PPC-PLA Block Copolymer Using One-Pot Two-Step Methodology*
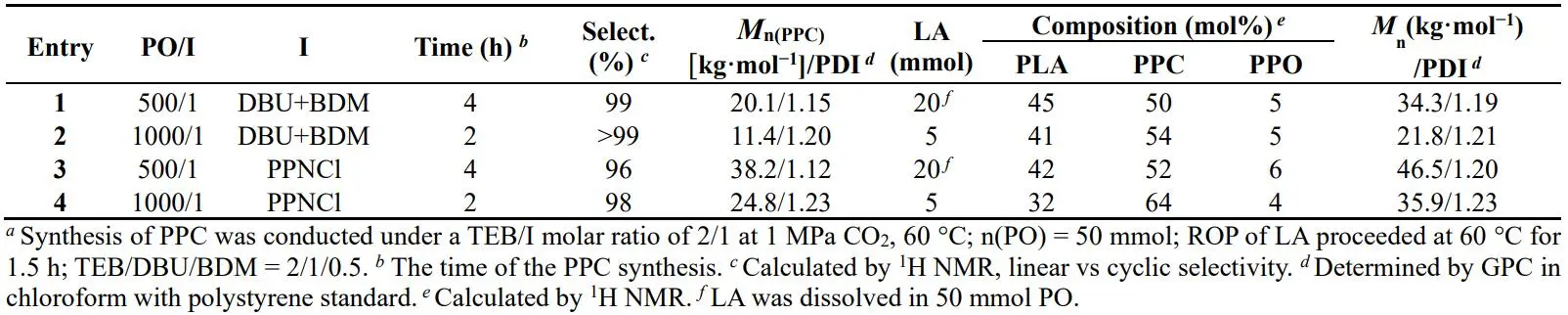
PPCLA has been successfully synthesized in one-pot one-step strategy catalyzed by TEB/bases. However, the reaction selectivity and the MWs (*M*
n < 15 kg·mol
−1) are not fully satisfied. So we changed the synthetic methodology to one-pot two-step strategy to afford the high selectivity and MW. shows the MWs and constitutions of the block copolymers obtained from different reaction conditions. Taking TEB/PPNCl catalyzed process as an example (, Entry 3, ), both Cl
− and trace water were initiators to prepare PPC with one active chain end and two active chain ends respectively, and then added LA into the reaction system to form PLA chain, wherein PPC acted as a macroinitiator to trigger the ROP of LA. As a result, PPC-PLA and PLA-PPC-PLA block copolymers were synthesized. The GPC peaks moving to the left represents that the MW of polymer increases with the continuous conversion of LA, which proves that the PLA block has been attached onto PPC chain. Via the two-step feeding, the MW increases from 38.2 kg·mol
−1 to 46.5 kg·mol
−1. The TEB/DBU pair displays the similar result but the target product is only PLA-PPC-PLA because no monofunctional initiator exists in the reaction system (, Entry 2, Figure S8). DOSY spectra just share a diffusion coefficient demonstrating the successful connection of PLA block to PPC chain (Figure S9).
. Synthesis of block copolymer catalyzed by different Lewis pairs in one-pot two-step strategy *a*.
. Synthesis of PPC-PLA block copolymers catalyzed by TEB/PPNCl: (a) evolution of <sup>1</sup>H NMR spectra of LA ROP with reaction time; (b) evolution of GPC traces and MWs.
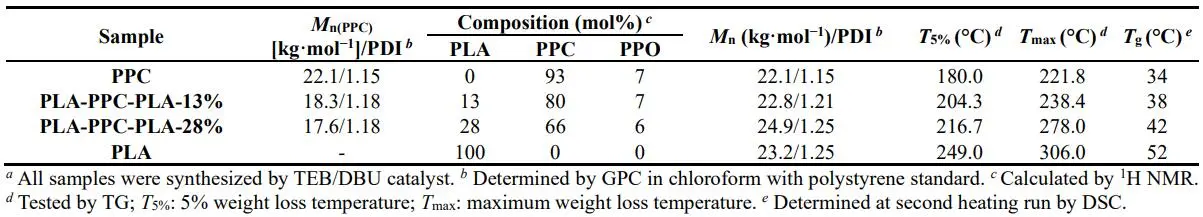
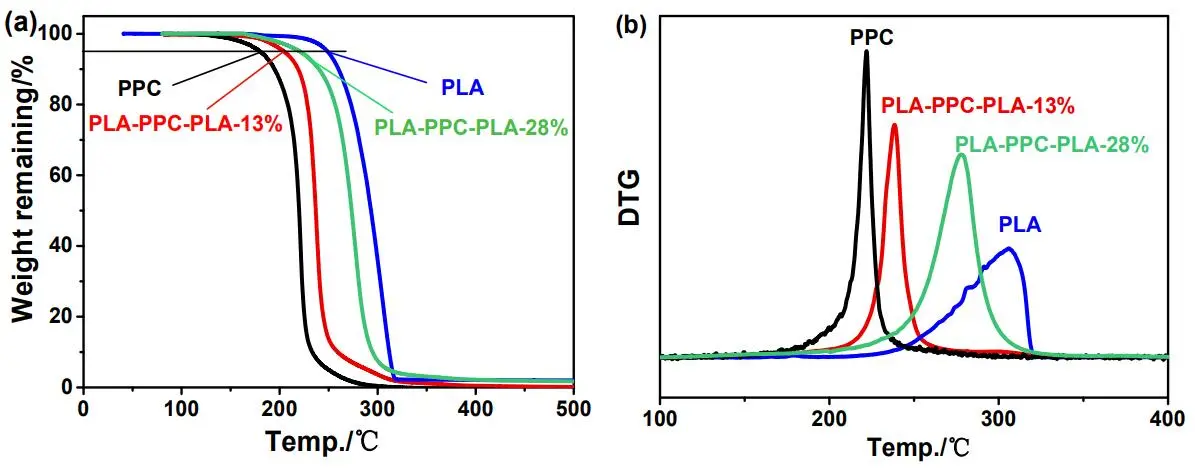
In order to compare the thermal stability of the triblock copolymers, PPC, PLA, PLA-PPC-PLA-13% and PLA-PPC-PLA-28% were synthesized by TEB/DBU catalyst (). Above four different polymers have almost the same MWs and PLA-PPC-PLA-13% has lower PLA content (13 mol%) than that of PLA-PPC-PLA-28% (28 mol%). Furthermore, thermal stability was evaluated via thermogravimetric analysis (). The maximum weight loss temperatures (*T*
maxs) of the two triblock copolymers appear between that of PPC (221.8 °C) and PLA (306.0 °C). High PLA content sample exhibits higher thermal stability. In addition, the triblock copolymers just display one glass transition temperature (*T*
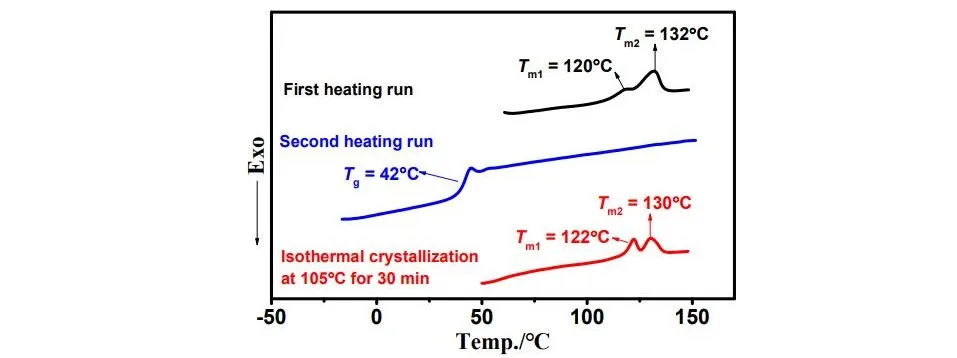
g) indicating good miscibility of PPC and PLA block. However, the melting peaks of PLA-PPC-PLA-13% and PLA-PPC-PLA-28% disappear in the second heating run of DSC test. As shown in , PLA-PPC-PLA-28% was heated in the first run of 10 °C/min, and then cooled at 10 °C/min rate, and finally heated for the second run at 10 °C/min. It displays *T*
ms at the first heating run and no melting peaks at the second heating run. This phenomenon is attributed to the low crystallization rate of PLA in PLA-PPC-PLA-28%. So we annealed the sample at 105 °C for 30 min after the first heating run, ensuring enough time for PLA-PPC-PLA-28% to crystallize. As excepted, two melting peaks (*T*
m1 = 122 °C, *T*
m2 = 130 °C) appear with a more distinct shape than that recorded at the first heating run. The crystalline characteristics correspond to the tri-block structure of PLA-PPC-PLA. What’s more, it’s common to find that PLA’s melting peak splits into two peaks corresponding to different crystal types, where the Tm of α crystal is 10 °C higher than that of β crystal [
12].
. Thermal properties of different samples *a*.
. (a) TG and (b) DTG curves of samples ().
. Three DSC curves of PLA-PPC-PLA-2 recorded at a 10 °C/min heating rate.
4. Conclusions
In this work, we have demonstrated that the ROP of LA and the terpolymerization of PO/LA/CO2 can successfully proceed using TEB/base Lewis pairs as the catalyst. In PLA synthesis catalyzed by TEB/PPNCl pair, PO was ring opened by Cl− to form PPN(OCH(CH3)CH2)Cl as the initiation step, which triggered the ROP of LA. The best ratio of TEB/PPNCl for PLA synthesis is 1/1. This ratio can effectively avoid the low reaction rate at high ratio and severe transesterification at low ratio. Then multiblock PPCLAs were synthesized using one-pot/one-step strategy. However, the MWs of resultant polymers were relatively low (*M*n < 15 kg·mol−1) even though the quaternary ammonium salt bases showed the better selectivity. In order to synthesize PLA-contained high-molecule-weight polymers, one-pot/two-step strategy was further adopted. PPC was first synthesized and then served as macroinitiator to initiate the ROP of LA without adding other catalyst to mediate the reaction. Consequently, PLA-PPC and PLA-PPC-PLA block copolymers were also synthesized from monofunctional and difunctional initiators respectively. PLA-PPC-PLA triblock copolymers displayed superior thermal stability compared with PPC, demonstrating that the introduction of PLA segments can effectively improve the thermal stability of PPC.
Supplementary Materials
Summarize the supplementary information with the caption names in this section. The following supporting information can be found at: https://www.sciepublish.com/index/journals/article/spe/30.html/id/14.
Author Contributions
Conceptualization, Z.H.; Methodology, D.H.; Software, S.Y.; Validation, J.L.; Formal Analysis, J.L.; Investigation, S.Y.; Resources, Y.M.; Data Curation, Y.R.; Writing – Original Draft Preparation, S.Y.; Writing – Review & Editing, M.X. and Y.M.; Visualization, Y.R.; Supervision, S.W.; Project Administration, Y.M.; Funding Acquisition, M.X. and Y.M.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Funding
This research was funded by the National Natural Science Foundation of China (Grant No. 51673131), the Fundamental Research Funds for the Central Universities (171gjc37), the financial supports from industrials: Shandong Lecron Industrial Development Group, Co., Ltd, China, Guangdong Tianxin New Material Technology Co., Ltd, China, Hebei CNC Risun Energy Co., Ltd, China and Huanghua Xinnuolixing Fine Chemicalstock Co., Ltd. China.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
1.
Zhang X, Fevre M, Jones GO, Waymouth RM. Catalysis as an enabling science for sustainable polymers.
Chem. Rev. 2018,
118, 839–885.
[Google Scholar]
-
2.
Tang S, Nozaki K. Advances in the synthesis of copolymers from carbon dioxide, dienes, and olefins.
Acc. Chem. Res. 2022,
55, 1524–1532.
[Google Scholar]
-
3.
Bhat GA, Darensbourg DJ. Progress in the catalytic reactions of CO
2 and epoxides to selectively provide cyclic or polymeric carbonates.
Green Chem. 2022,
24, 5007–5034.
[Google Scholar]
-
4.
Wu J, Yu TL, Chen CT, Lin CC. Recent developments in main group metal complexes catalyzed/initiated polymerization of lactides and related cyclic esters.
Coord. Chem. Rev. 2006,
250, 602–626.
[Google Scholar]
-
5.
Xu Y, Lin L, Xiao M, Wang S, Smith AT, Sun L, et al. Synthesis and properties of CO
2-based plastics: Environmentally-friendly, energy-saving and biomedical polymeric materials.
Prog. Polym. Sci. 2018,
80, 163–182.
[Google Scholar]
-
6.
Nampoothiri KM, Nair NR, John RP. An overview of the recent developments in polylactide (PLA) research.
Bioresour. Technol. 2010,
101, 8493–8501.
[Google Scholar]
-
7.
Rasal RM, Janorkar AV, Hirt DE. Poly(lactic acid) modifications.
Prog. Polym. Sci. 2010,
35, 338–356.
[Google Scholar]
-
8.
Hamad K, Kaseem M, Ayyoob M, Joo J, Deri F. Polylactic acid blends: The future of green, light and tough.
Prog. Polym. Sci. 2018,
85, 83–127.
[Google Scholar]
-
9.
Bednarek M. Branched aliphatic polyesters by ring-opening (co)polymerization.
Prog. Polym. Sci. 2016,
58, 27–58.
[Google Scholar]
-
10.
Luinstra G. Poly(propylene carbonate), old copolymers of propylene oxide and carbon dioxide with new interests: Catalysis and material properties.
Polym. Rev. 2008,
48, 192–219.
[Google Scholar]
-
11.
Liu SQ, Wang JL, Huang KL, Liu YF, Wu WK. Synthesis of poly(propylene-co-lactide carbonate) and hydrolysis of the terpolymer.
Polym. Bull. 2011,
66, 327–340.
[Google Scholar]
-
12.
Tang L, Luo W, Xiao M, Wang S, Meng Y. One-pot synthesis of terpolymers with long ʟ-lactide rich sequence derived from propylene oxide, CO
2, and ʟ-lactide catalyzed by zinc adipate.
J. Polym. Sci. A Polym. Chem. 2015,
53, 1734–1741.
[Google Scholar]
-
13.
Song P, Xu H, Mao X, Liu X, Wang L. A one-step strategy for aliphatic poly(carbonate-ester)s with high performance derived from CO
2, propylene oxide and ʟ-lactide.
Polym. Adv. Technol. 2017,
28, 736–741.
[Google Scholar]
-
14.
Xie D, Yang Z, Wu L, Zhang C, Chisholm MH. One-pot regioselective and stereoselective terpolymerization of rac-lactide, CO
2 and rac-propylene oxide with TPPMCl (M = Cr, Co, Al)/PPNCl binary catalyst.
Polym. Int. 2018,
67, 883–893.
[Google Scholar]
-
15.
Li X, Hu C, Pang X, Duan R, Chen X. One-pot copolymerization of epoxides/carbon dioxide and lactide using a ternary catalyst system.
Catal. Sci. Technol. 2018,
8, 6452–6457.
[Google Scholar]
-
16.
Duan R, Hu C, Sun Z, Zhang H, Pang X, Chen X. Conjugated tri-nuclear salen-Co complexes for the copolymerization of epoxides/CO
2: Cocatalyst-free catalysis.
Green Chem. 2019,
21, 4723–4731.
[Google Scholar]
-
17.
Wu GP, Darensbourg DJ, Lu XB. Tandem metal-coordination copolymerization and organocatalytic ring-opening polymerization via water to synthesize diblock copolymers of styrene oxide/CO
2 and lactide.
J. Am. Chem. Soc. 2012,
134, 17739–17745.
[Google Scholar]
-
18.
Lohmeijer BGG, Pratt RC, Leibfarth F, Logan JW, Hedrick JL. Guanidine and amidine organocatalysts for ring-opening polymerization of cyclic esters.
Macromolecules 2006,
39, 8574–8583.
[Google Scholar]
-
19.
Darensbourg DJ, Wu G. A one-pot synthesis of a triblock copolymer from propylene oxide/carbon dioxide and lactide: Intermediacy of polyol initiators.
Angew. Chem. Int. Ed. 2013,
52, 10602–10606.
[Google Scholar]
-
20.
Wu GP, Darensbourg DJ. Mechanistic insights into water-mediated tandem catalysis of metal-coordination CO
2/epoxide copolymerization and organocatalytic ring-opening polymerization: One-pot, two steps, and three catalysis cycles for triblock copolymers synthesis.
Macromolecules 2016,
49, 807–814.
[Google Scholar]
-
21.
Hu C, Duan R, Yang S, Pang X, Chen X. CO
2 controlled catalysis: Switchable homopolymerization and copolymerization.
Macromolecules 2018,
51, 4699–4704.
[Google Scholar]
-
22.
Zhang D, Boopathi SK, Hadjichristidis N, Gnanou Y, Feng X. Metal-free alternating copolymerization of CO
2 with epoxides: Fulfilling “green” synthesis and activity.
J. Am. Chem. Soc. 2016,
138, 11117–111120.
[Google Scholar]
-
23.
Wang Y, Zhang JY, Yang JL, Zhang HK, Kiriratnikom J, Zhang CJ, et al. Highly selective and productive synthesis of a carbon dioxide-based copolymer upon zwitterionic growth.
Macromolecules 2021,
54, 2178–2186.
[Google Scholar]
-
24.
Chen Z, Yang JL, Lu XY, Hu LF, Cao XH, Wu GP, et al. Triethyl borane-regulated selective production of polycarbonates and cyclic carbonates for the coupling reaction of CO
2 with epoxides.
Polym. Chem. 2019,
10, 3621–3628.
[Google Scholar]
-
25.
Patil NG, Boopathi SK, Alagi P, Hadjichristidis N, Gnanou Y, Feng X. Carboxylate salts as ideal initiators for the metal-free copolymerization of CO
2 with epoxides: Synthesis of well-defined polycarbonates diols and polyols.
Macromolecules 2019,
52, 2431–2438.
[Google Scholar]
-
26.
Papagiannopoulos A, Zhao J, Zhang G, Pispas S, Jafta CJ. Viscosity transitions driven by thermoresponsive self-assembly in PHOS-g-P(PO-r-EO) brush copolymer.
Macromolecules 2018,
51, 1644–1653.
[Google Scholar]
-
27.
Chen Y, Shen J, Liu S, Zhao J, Wang Y, Zhang G. High efficiency organic Lewis pair catalyst for ring-opening polymerization of epoxides with chemoselectivity.
Macromolecules 2018,
51, 8286–8297.
[Google Scholar]
-
28.
Zhang C, Duan H, Hu L, Zhang C, Zhang X. Metal-free route to precise synthesis of poly(propylene oxide) and its blocks with high activity.
ChemSusChem 2018,
11, 4209–4213.
[Google Scholar]
-
29.
Boopathi SK, Hadjichristidis N, Gnanou Y, Feng X. Direct access to poly(glycidyl azide) and its copolymers through anionic (co-)polymerization of glycidyl azide.
Nat. Commun. 2019,
10, 293.
[Google Scholar]
-
30.
Hu L, Zhang C, Wu H, Yang J, Liu B, Duan H, et al. Highly active organic Lewis pairs for the copolymerization of epoxides with cyclic anhydrides: Metal-free access to well-defined aliphatic polyesters.
Macromolecules 2018,
51, 3126–3134.
[Google Scholar]
-
31.
Zhu S, Wang Y, Ding W, Zhou X, Liao Y, Xie X. Lewis pair catalyzed highly selective polymerization for the one-step synthesis of AzCy(AB)xCyAz pentablock terpolymers.
Polym. Chem. 2020,
11, 1691–1695.
[Google Scholar]
-
32.
Kummari A, Pappuru S, Chakraborty D. Fully alternating and regioselective ring-opening copolymerization of phthalic anhydride with epoxides using highly active metal-free Lewis pairs as a catalyst.
Polym. Chem. 2018,
9, 4052–4062.
[Google Scholar]
-
33.
Zhang B, Li H, Luo H, Zhao J. Ring-opening alternating copolymerization of epichlorohydrin and cyclic anhydrides using single- and two-component metal-free catalysts.
Eur. Polym. J. 2020,
134, 109820.
[Google Scholar]
-
34.
Ji HY, Chen XL, Wang B, Pan L, Li YS. Metal-free, regioselective and stereoregular alternating copolymerization of monosubstituted epoxides and tricyclic anhydrides.
Green Chem. 2018,
20, 3963–3973.
[Google Scholar]
-
35.
Li H, He G, Chen Y, Zhao J, Zhang G. One-step approach to polyester–polyether block copolymers using highly tunable bicomponent catalyst.
ACS Macro Lett. 2019,
8, 973–978.
[Google Scholar]
-
36.
Ji HY, Song DP, Wang B, Pan L, Li YS. Organic Lewis pairs for selective copolymerization of epoxides with anhydrides to access sequence-controlled block copolymers.
Green Chem. 2019,
21, 6123–6132.
[Google Scholar]
-
37.
Liu S, Bai T, Ni K, Chen Y, Zhao J, Ling J, et al. Biased Lewis pair: A general catalytic approach to ether-ester block copolymers with unlimited sequences.
Angew. Chem. Int. Ed. 2019,
58, 15478–15487.
[Google Scholar]
-
38.
Varghese JK, Hadjichristidis N, Gnanou Y, Feng X. Degradable poly(ethylene oxide) through metal-free copolymerization of ethylene oxide with ʟ-lactide.
Polym. Chem. 2019,
10, 3764–3771.
[Google Scholar]
-
39.
Chidara VK, Boopathi SK, Hadjichristidis N, Gnanou Y, Feng X. Triethylborane-assisted synthesis of random and block poly(ester-carbonate)s through one-pot terpolymerization of epoxides, CO
2, and cyclic anhydrides.
Macromolecules 2021,
54, 2711–2719.
[Google Scholar]
-
40.
Liang J, Ye S, Wang W, Fan C, Wang S, Han D, et al. Performance tailorable terpolymers synthesized from carbon dioxide, phthalic anhydride and propylene oxide using Lewis acid-base dual catalysts.
J. CO2 Util. 2021,
49, 101558.
[Google Scholar]
-
41.
Ye S, Wang W, Liang J, Wang S, Xiao M, Meng Y. Metal-free approach for a one-pot construction of biodegradable block copolymers from epoxides, phthalic anhydride, and CO
2.
ACS Sustain. Chem. Eng. 2020,
8, 17860–17867.
[Google Scholar]
-
42.
Zhang J, Wang L, Liu S, Kang X, Li Z. A Lewis pair as organocatalyst for one-pot synthesis of block copolymers from a mixture of epoxide, anhydride, and CO
2.
Macromolecules 2021,
54, 763–772.
[Google Scholar]
-
43.
Yang J, Wu H, Li Y, Zhang X, Darensbourg DJ. Perfectly alternating and regioselective copolymerization of carbonyl sulfide and epoxides by metal-free Lewis pairs.
Angew. Chem. Int. Ed. 2017,
56, 5774–5779.
[Google Scholar]
-
44.
Hošťálek Z, Trhlíková O, Walterová Z, Martinez T, Peruch F, Cramail H, et al. Alternating copolymerization of epoxides with anhydrides initiated by organic bases.
Eur. Polym. J. 2017,
88, 433–447.
[Google Scholar]
-
45.
Zhang D, Feng X, Gnanou Y, Huang K. Theoretical mechanistic investigation into metal-free alternating copolymerization of CO
2 and epoxides: The key role of triethylborane.
Macromolecules 2018,
51, 5600–5607.
[Google Scholar]