PIEZO Mechanotransduction in the Cardiovascular System: Physiological Roles and Disease Implications

PIEZO Mechanotransduction in the Cardiovascular System: Physiological Roles and Disease Implications
Jun Xu *
Received: 16 May 2026 Revised: 27 May 2026 Accepted: 02 June 2026 Published: 12 June 2026

© 2026 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
1. Introduction
Mechanical forces are fundamental regulators of cardiovascular physiology and pathology. Throughout the cardiovascular system, cells are continuously exposed to diverse biomechanical stimuli, including shear stress generated by blood flow, cyclic stretch during cardiac contraction, hydrostatic pressure, and extracellular matrix stiffness [1,2]. To maintain tissue homeostasis and adapt to changing hemodynamic conditions, cardiovascular cells must possess efficient mechanotransduction systems capable of converting mechanical inputs into biochemical and electrophysiological signals. Mechanotransduction therefore, represents a central mechanism governing vascular tone, endothelial integrity, myocardial remodeling, inflammation, and cellular adaptation to stress [3,4].
Mechanotransduction in cardiovascular tissues is mediated through highly integrated mechanosensory systems that collectively regulate vascular homeostasis, myocardial remodeling, inflammatory activation, and tissue adaptation under mechanical stress. In addition to mechanosensitive ion channels, cardiovascular cells rely on integrin-mediated focal adhesion signaling, cytoskeletal force transmission, extracellular matrix interactions, and transcriptional mechanoregulators such as YAP/TAZ to sense and respond to biomechanical forces. Integrin–focal adhesion kinase (FAK) signaling pathways play essential roles in endothelial adaptation, vascular remodeling, and cardiac hypertrophy under hemodynamic loading conditions [5,6]. Similarly, YAP/TAZ-mediated mechanotransduction dynamically responds to matrix stiffness and cytoskeletal tension to regulate fibrosis, proliferation, and remodeling [7]. Transient receptor potential (TRP) channels, particularly TRPV4 and TRPC6, additionally contribute to calcium-dependent mechanosensitive signaling in endothelial cells, cardiomyocytes, and vascular smooth muscle cells [8,9,10]. PIEZO channels, therefore, function within a broader interconnected mechanosensory network rather than as isolated mechanotransducers.
Mechanosensitive ion channels are among the most rapid and direct mediators of mechanotransduction because they can respond within milliseconds to membrane deformation and mechanical loading [11]. Among mechanosensitive ion channels, PIEZO1 and PIEZO2 have emerged as critical mechanosensors in mammalian tissues. The discovery of PIEZO proteins by Coste and colleagues was a landmark advancement in mechanobiology, establishing the molecular basis for mechanically activated ion channels in mammalian systems [12]. Their seminal work demonstrated that heterologous expression of PIEZO proteins generated mechanically activated cation currents in otherwise mechanically insensitive cells, thereby identifying PIEZO proteins as pore-forming mechanosensitive ion channels directly involved in cellular force sensing. This discovery fundamentally transformed the understanding of mechanotransduction and initiated substantial expansion of mechanobiology research across cardiovascular, sensory, and developmental systems.
Subsequent studies demonstrated that PIEZO channels are intrinsically mechanosensitive and can be directly gated by membrane tension through a “force-from-lipids” mechanism [13,14]. Structural studies using cryo-electron microscopy further revealed their unique curved, propeller-like architecture that enables membrane deformation and mechanogating [15,16,17]. Mechanical stimuli such as shear stress or stretch flatten the membrane dome surrounding the channel, inducing conformational changes that open the ion-conducting pore [18,19]. Additional studies have shown that PIEZO gating is influenced by cytoskeletal interactions, membrane lipid composition, voltage dependence, and extracellular matrix mechanics [20,21,22,23].
Although PIEZO channels were first investigated primarily in sensory neurons involved in touch and proprioception [24,25], growing evidence indicates that PIEZO-mediated mechanotransduction plays a pivotal role in cardiovascular biology [26,27,28]. PIEZO1 is highly expressed in endothelial cells, cardiomyocytes, fibroblasts, erythrocytes, and vascular smooth muscle cells, where it mediates responses to fluid shear stress, stretch, and mechanical deformation [29,30,31,32]. Endothelial PIEZO1 regulates vascular development, nitric oxide signaling, ATP release, and blood pressure homeostasis [24,30]. In the myocardium, PIEZO1 contributes to mechano-chemo transduction, calcium handling, hypertrophic signaling, and remodeling responses under pressure overload [33,34,35]. In cardiac fibroblasts, PIEZO1 activation promotes inflammatory and profibrotic signaling pathways associated with extracellular matrix remodeling and fibrosis [36,37,38].
Dysregulation of PIEZO signaling has increasingly been implicated in cardiovascular disease. Abnormal PIEZO-mediated mechanotransduction contributes to hypertension, endothelial dysfunction, cardiac hypertrophy, ischemic injury, fibrosis, vascular inflammation, and heart failure progression [39,40,41]. For example, enhanced PIEZO1 activity has been associated with pathological calcium influx and activation of calpain/calcineurin signaling pathways that promote maladaptive hypertrophy [39]. Similarly, disturbed shear stress can induce PIEZO1-mediated inflammatory activation of circulating monocytes and vascular endothelial cells, linking mechanotransduction to atherosclerosis and vascular remodeling [42]. Emerging evidence also suggests important roles for PIEZO channels in peripheral arterial disease, lymphatic remodeling, and ischemia-induced adverse cardiac remodeling [40,43,44].
Recent advances in structural biology, molecular genetics, electrophysiology, and computational modeling have greatly accelerated understanding of PIEZO channel function. In particular, artificial intelligence and machine learning approaches are beginning to provide new tools for analyzing ion channel kinetics and mechanosensitive electrophysiological data. Deep learning architectures such as recurrent convolutional neural networks and long short-term memory models have demonstrated the potential to identify hidden gating states and transition kinetics from noisy patch-clamp recordings, thereby offering new opportunities to understand the complex dynamics of PIEZO mechanotransduction.
This review synthesizes current understanding of PIEZO mechanotransduction in the cardiovascular system, focusing on structural and biophysical mechanisms, physiological functions, disease implications, and emerging therapeutic opportunities. Particular emphasis is placed on recent advances in cardiovascular mechanobiology and computational approaches that may facilitate future translational applications of PIEZO-targeted therapies.
2. Structural and Biophysical Basis of PIEZO Mechanotransduction
2.1. Discovery and Molecular Architecture
The discovery of PIEZO proteins represented a landmark advancement in the field of mechanobiology and significantly transformed the understanding of how cells detect and respond to mechanical stimuli. Prior to their identification, the molecular basis of mammalian mechanotransduction remained largely elusive despite extensive evidence demonstrating that mechanical forces regulate numerous physiological processes, including vascular homeostasis, touch sensation, hearing, and cardiac adaptation [1,45]. In 2010–2012, Coste and colleagues identified PIEZO1 and PIEZO2 as a previously unknown family of mechanically activated ion channels that directly convert membrane deformation into ionic currents [46,47]. Functional studies demonstrated that heterologous expression of PIEZO proteins generated mechanically activated currents in otherwise mechanically insensitive cells, establishing PIEZO proteins as pore-forming mechanosensitive ion channels [47].
Prior to the identification of PIEZO channels, the molecular mechanisms governing mammalian mechanotransduction remained poorly understood despite extensive evidence that biomechanical forces regulate cardiovascular physiology and disease [6]. The discovery of PIEZO proteins therefore provided a direct mechanistic link between membrane deformation and ion channel activation, significantly advancing the broader field of cardiovascular mechanobiology.
PIEZO1 and PIEZO2 are exceptionally large transmembrane proteins consisting of more than 2500 amino acids and containing over 30 transmembrane helices per subunit, making them among the largest ion channels identified in mammalian systems [48,49]. Structural analyses revealed that three identical subunits assemble into a homotrimeric complex with a distinctive three-bladed propeller-like geometry surrounding a centrally located ion-conducting pore [15]. Each blade extends outward into the lipid bilayer through long curved peripheral arms that impart substantial membrane curvature. This unusual architecture distinguishes PIEZO channels from conventional ligand-gated or voltage-gated ion channels and is central to their mechanosensory function.
Breakthrough cryo-electron microscopy studies provided high-resolution structural insights into PIEZO channel organization and mechanogating mechanisms [16,17]. These studies demonstrated that PIEZO channels deform the surrounding lipid bilayer into a curved dome-like structure, often referred to as a “nano-bowl” configuration. The membrane curvature generated by PIEZO channels is believed to store elastic energy that contributes directly to mechanosensitivity. The central pore module is surrounded by intracellular beam-like structures and extracellular cap domains that mechanically couple peripheral membrane deformation to pore opening [17,32].
Mechanical activation of PIEZO channels is strongly linked to membrane tension and curvature. Structural and biophysical investigations showed that external mechanical forces such as shear stress, stretch, pressure, or membrane indentation flatten the curved membrane dome and induce conformational rearrangements within the peripheral blades and beam structures [18,50]. These force-induced structural changes propagate toward the central pore region, ultimately triggering channel opening and allowing the influx of cations, particularly calcium ions. Because calcium functions as a major intracellular second messenger, PIEZO activation rapidly initiates downstream signaling pathways involved in mechanotransduction, gene regulation, cytoskeletal remodeling, and cellular adaptation [27,45,51,52].
Figure 1 illustrates the structural architecture and mechanogating mechanism of PIEZO channels. Figure 1A presents the overall trimeric organization of the PIEZO channel embedded within the lipid bilayer. Each subunit forms one of the three curved peripheral blades surrounding a central ion-conducting pore, creating the characteristic propeller-like structure observed in cryo-electron microscopy studies. Figure 1B depicts the membrane dome configuration generated by the intrinsic curvature of the PIEZO complex within the lipid bilayer. This dome-shaped deformation enables the channel to function as a nanoscale mechanosensor that detects membrane tension and curvature changes. Figure 1C illustrates the beam structures that extend from the peripheral blades toward the central pore region. These beam-like intracellular elements act as mechanical transducers, transmitting force-induced conformational changes across the channel structure. Figure 1D demonstrates the force-from-lipids mechanogating mechanism. Under resting conditions, the membrane dome remains curved; however, mechanical forces such as shear stress, stretch, or membrane tension flatten the dome structure, propagating structural deformation through the beam elements toward the central pore. This conformational rearrangement results in pore opening and calcium-permeable cation influx, thereby initiating downstream mechanotransduction signaling pathways involved in cardiovascular physiology and disease.
In addition to mechanical regulation, PIEZO channels exhibit complex multimodal gating properties. Electrophysiological studies revealed that membrane voltage influences PIEZO channel activation and inactivation kinetics, suggesting important interactions between mechanical and electrical stimuli [22]. Recent investigations have also demonstrated rapid inactivation behavior, stochastic single-channel gating, and spring-like mechanical responses that enable PIEZO channels to respond dynamically to transient mechanical forces [53]. These kinetic properties are particularly important in cardiovascular tissues, where cells are continuously exposed to pulsatile flow and cyclic mechanical loading.
Collectively, structural and biophysical studies have established PIEZO channels as highly specialized membrane-embedded mechanotransducers optimized for sensing mechanical deformation at the nanoscale. Their unique architecture, intrinsic mechanosensitivity, and direct coupling between membrane mechanics and ion permeation provide the molecular foundation for their diverse physiological functions in the cardiovascular system and other mechanically active tissues.
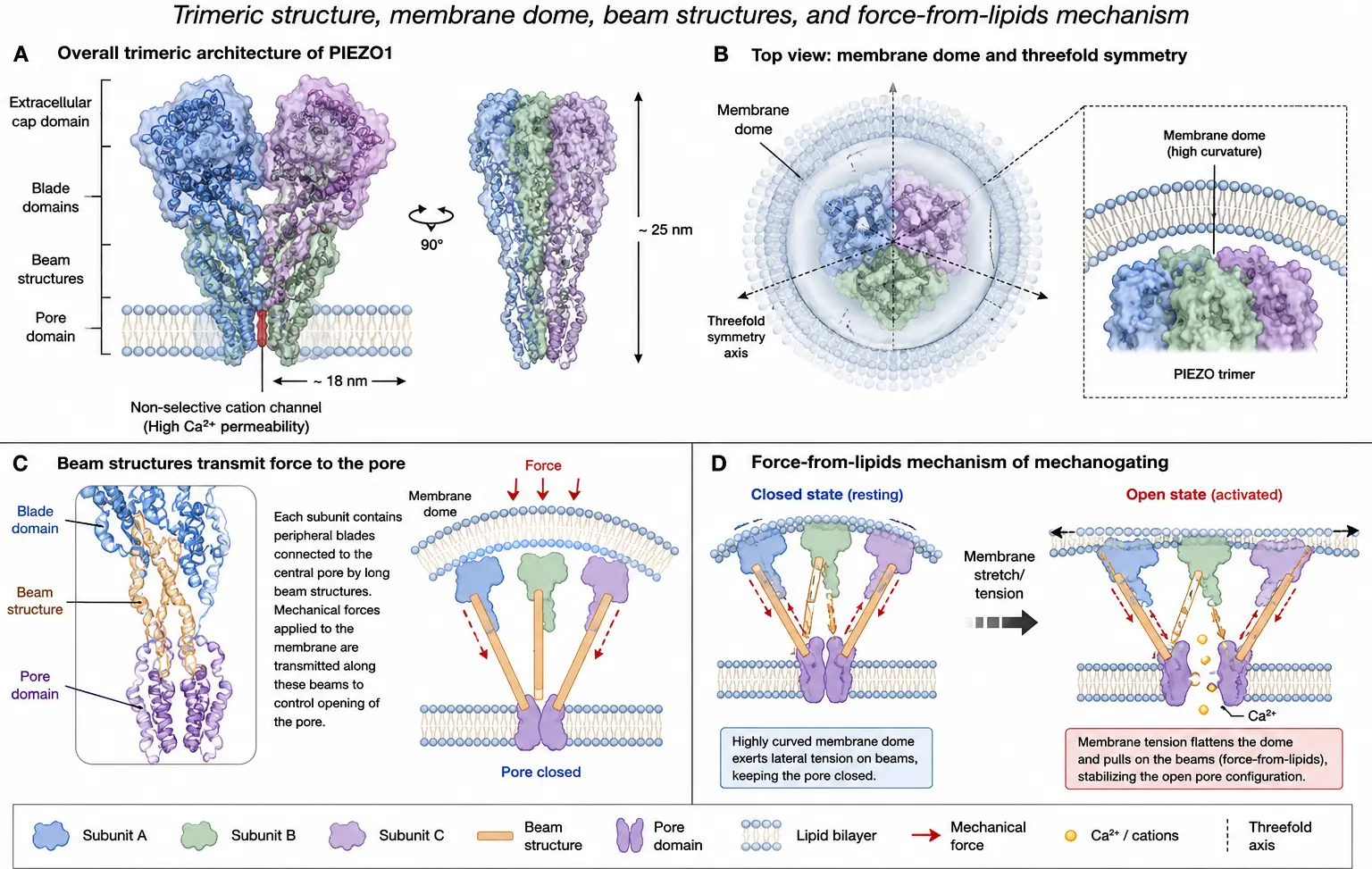
Figure 1. PIEZO structural architecture and mechanogating mechanism. (A) Overall trimeric architecture of the PIEZO channel embedded within the lipid bilayer, showing the characteristic propeller-like organization surrounding the central ion-conducting pore. (B) Dome-shaped membrane curvature generated by the PIEZO complex, enabling mechanosensation through membrane deformation. (C) Beam structures that mechanically couple the peripheral blades to the central pore region and transmit force-induced conformational changes. (D) Schematic illustration of the force-from-lipids mechanogating mechanism, in which membrane tension, shear stress, or mechanical stretch flattens the membrane dome, induces structural rearrangement, opens the central pore, and permits calcium-permeable cation influx to initiate downstream mechanotransduction signaling.
2.2. Mechanogating and Ion Permeation
PIEZO channels function as non-selective mechanosensitive cation channels with significant permeability to calcium ions, thereby enabling rapid conversion of mechanical stimuli into intracellular biochemical signaling events [54,55]. Upon activation, PIEZO-mediated calcium influx initiates multiple downstream signaling pathways involved in cytoskeletal remodeling, gene transcription, inflammatory activation, cellular adaptation, and mechanotransduction-dependent physiological responses [27,56]. In cardiovascular tissues, these calcium-dependent signaling cascades are particularly important for regulating endothelial nitric oxide production, vascular tone, cardiac hypertrophy, fibroblast activation, and mechano-chemo coupling [30,31,39].
Electrophysiological studies have demonstrated that PIEZO channels possess exceptionally rapid activation and inactivation kinetics, allowing them to respond dynamically to transient and repetitive mechanical forces [55,57]. These kinetic properties are essential in mechanically active tissues such as the cardiovascular system, where cells experience pulsatile flow, cyclic stretch, and pressure oscillations on a beat-to-beat basis. Rapid activation enables efficient sensing of abrupt mechanical perturbations, whereas rapid inactivation prevents excessive calcium loading and cellular toxicity during prolonged stimulation [58,59]. Recent studies further suggest that spring-like mechanical elements within the channel structure contribute to rapid inactivation and stochastic gating behavior [53].
Experimental studies using purified PIEZO1 channels reconstituted into artificial lipid bilayers demonstrated that membrane force alone is sufficient for activation, confirming intrinsic mechanosensitivity of the channel [60]. Nevertheless, subsequent studies revealed that cellular context significantly modulates PIEZO sensitivity and gating behavior. Cytoskeletal interactions can either stabilize membrane architecture or mechanically shield the channel, thereby influencing activation thresholds and channel kinetics [21,61]. Disruption of cytoskeletal elements such as actin filaments increases PIEZO1 mechanosensitivity by removing mechanoprotective constraints on the membrane [21].
Membrane lipid composition also plays a major role in regulating PIEZO activity. Variations in cholesterol concentration, phospholipid organization, and membrane stiffness can substantially alter mechanosensitivity by modifying local membrane curvature, elasticity, and tension propagation [20,62]. Cholesterol-rich membrane domains may stiffen the lipid bilayer and alter the energy required for channel opening, whereas changes in phospholipid saturation can affect membrane fluidity and force transmission. These findings indicate that PIEZO channels function not only as mechanosensors but also as integrators of the physical properties of the surrounding membrane environment.
Extracellular matrix stiffness and substrate mechanics further influence PIEZO channel activation, particularly in cardiovascular tissues undergoing remodeling and fibrosis [3]. Increased matrix stiffness enhances mechanical force transmission to the plasma membrane and may amplify PIEZO-mediated signaling in endothelial cells, cardiomyocytes, and fibroblasts. Such interactions are highly relevant in pathological conditions, including hypertension, cardiac hypertrophy, and fibrotic remodeling, where tissue stiffening progressively alters mechanotransduction dynamics [37,38].
In addition to mechanical gating, PIEZO channels exhibit multimodal regulation involving membrane voltage and chemical signaling pathways. Moroni et al. [22] demonstrated that PIEZO channels display voltage-dependent gating properties, indicating that membrane potential can modulate activation and inactivation kinetics. This voltage sensitivity suggests functional crosstalk between electrical and mechanical stimuli, particularly in excitable tissues such as the heart and vasculature. Regulatory pathways involving phosphorylation, intracellular calcium, inflammatory mediators, and membrane-associated proteins have also been shown to influence PIEZO channel function [23,63].
Recent studies have additionally highlighted the complexity of PIEZO ion permeation behavior. Although PIEZO channels are classically described as non-selective cation channels, emerging evidence suggests context-dependent ion selectivity and rectification properties [64]. Structural studies indicate that pore geometry, electrostatic interactions, and conformational states contribute to ion permeation characteristics and gating efficiency [31,32,54]. Continued investigation of these mechanisms remains essential for understanding how PIEZO channels regulate mechanotransduction under physiological and pathological conditions.
Overall, PIEZO mechanogating represents a highly integrated process involving membrane tension sensing, structural deformation, pore opening, and calcium-mediated signal transduction. The dynamic interplay between channel structure, membrane mechanics, cytoskeletal organization, and extracellular environment enables PIEZO channels to function as sophisticated biomechanical transducers within the cardiovascular system and other mechanically active tissues.
2.3. PIEZO Channel Kinetics and Computational Modeling
Understanding the gating kinetics of PIEZO channels remains one of the most important and challenging areas in mechanobiology because mechanosensitive ion channel behavior is highly dynamic, nonlinear, and stochastic in nature [55,57]. Unlike many classical ligand-gated or voltage-gated ion channels, PIEZO channels respond directly to rapidly changing mechanical forces such as membrane tension, shear stress, indentation, and cyclic stretch, producing transient currents with complex activation and inactivation dynamics [19,58]. These properties are particularly important in cardiovascular tissues, where endothelial cells, cardiomyocytes, fibroblasts, and vascular smooth muscle cells experience continuously fluctuating biomechanical environments. Consequently, accurate characterization of PIEZO channel kinetics is essential for understanding cardiovascular mechanotransduction under both physiological and pathological conditions.
Electrophysiological studies using patch-clamp techniques have provided important insights into PIEZO channel behavior at both whole-cell and single-channel levels [65]. Patch-clamp electrophysiology enables high-resolution recording of mechanically activated ionic currents and allows investigators to quantify channel conductance, activation thresholds, inactivation kinetics, ion permeation properties, and force-response relationships [55]. Using pressure-clamp or mechanical indentation protocols, researchers have demonstrated that PIEZO channels exhibit rapid activation followed by fast inactivation, with kinetic behavior strongly influenced by membrane tension, cytoskeletal organization, lipid composition, and voltage dependence [22,59].
Despite major experimental advances, interpretation of PIEZO channel kinetics remains computationally difficult because channel activity often involves hidden conformational states, stochastic transitions, and substantial experimental noise [66,67]. Mechanically activated ion channels can display multiple subconductance states, variable transition probabilities, and force-dependent gating pathways that are challenging to characterize using traditional analytical methods based on deterministic curve fitting or manual hidden Markov modeling [55]. Moreover, the large volume of electrophysiological data generated from high-frequency recordings makes manual analysis time-consuming and susceptible to user bias.
Recent advances in computational modeling, artificial intelligence, and machine learning have therefore emerged as promising approaches for deciphering PIEZO channel kinetics and mechanotransduction dynamics. Deep learning methods are particularly attractive because they can identify complex nonlinear patterns and hidden state transitions within large, noisy datasets that are difficult to analyze using conventional statistical approaches. Inspired by these advances, computational frameworks utilizing recurrent convolutional neural networks (RCNNs), convolutional neural networks (CNNs), and long short-term memory (LSTM) architectures have been proposed for studying PIEZO channel kinetics and force-dependent gating behavior.
Figure 2 illustrates an artificial intelligence-assisted workflow for analyzing PIEZO channel electrophysiology and mechanotransduction dynamics. The workflow begins with patch-clamp electrophysiological recordings that capture mechanically activated ion channel currents with high temporal resolution. These recordings often contain substantial background noise and complex stochastic gating behavior, making conventional manual analysis difficult and time consuming. Following data acquisition, signal preprocessing techniques such as filtering, normalization, and feature extraction are applied to improve data quality and isolate relevant ion channel events. Deep learning architectures, including convolutional neural networks (CNNs), recurrent neural networks (RNNs), recurrent convolutional neural networks (RCNNs), and long short-term memory (LSTM) networks, are then used to analyze electrophysiological datasets and identify gating transitions, hidden kinetic states, and force-dependent channel behavior. Machine learning-based approaches enable improved detection of stochastic ion channel activity, prediction of transition probabilities, and reconstruction of mechanotransduction dynamics from noisy experimental recordings. The integration of electrophysiology, artificial intelligence, and computational modeling, therefore, provides a powerful framework for studying PIEZO channel kinetics, multiscale mechanobiology, AI-assisted drug discovery, and precision cardiovascular mechanotransduction research.
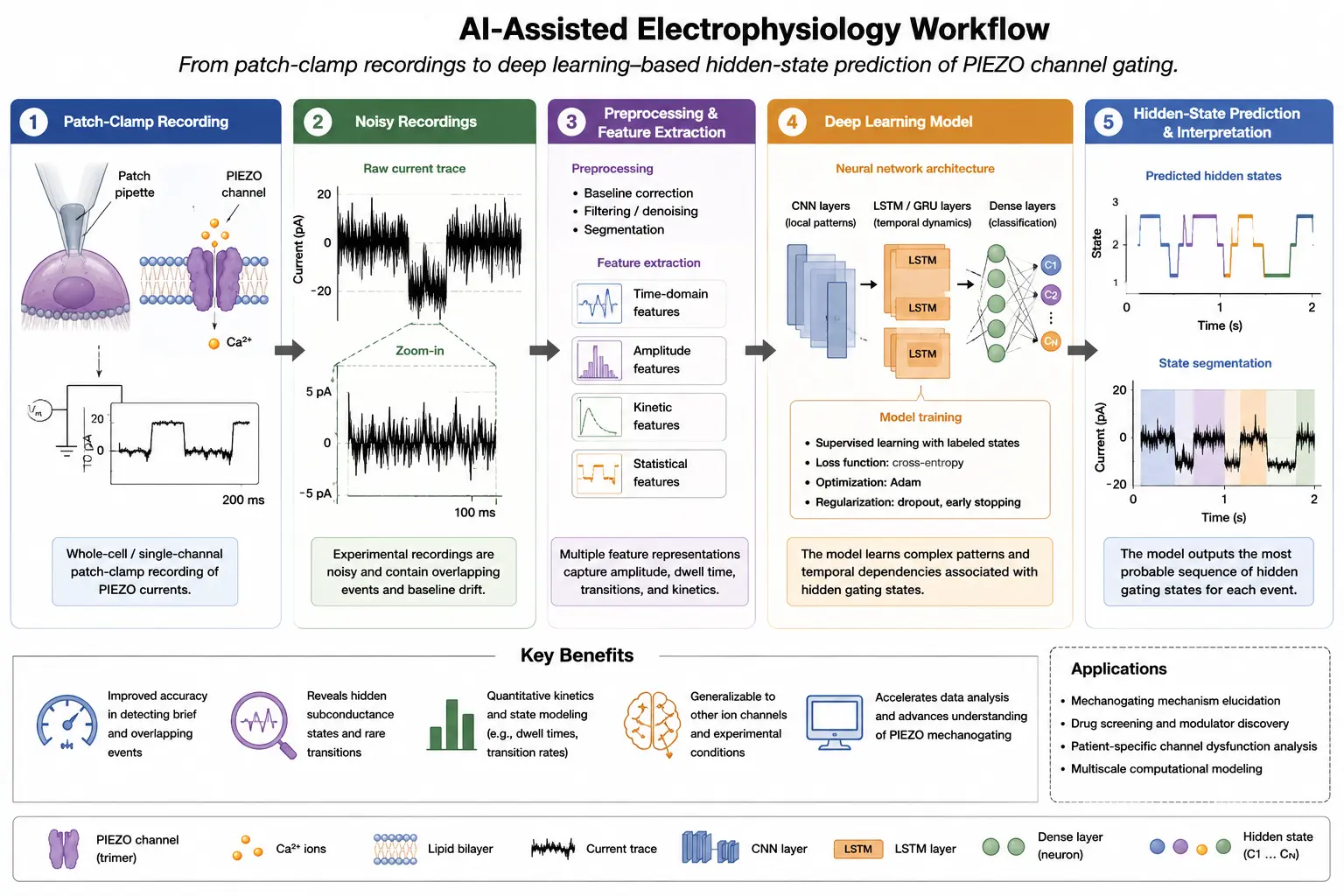
Figure 2. AI-assisted electrophysiology workflow for PIEZO channel kinetics analysis. Schematic illustration of an artificial intelligence-assisted workflow for analyzing mechanosensitive ion channel electrophysiology. Patch-clamp electrophysiological recordings containing noisy single-channel current traces are first acquired and preprocessed for signal filtering and feature extraction. Deep learning architectures, including convolutional neural networks (CNNs), recurrent neural networks (RNNs), recurrent convolutional neural networks (RCNNs), and long short-term memory (LSTM) networks, are then applied to identify gating events, hidden kinetic states, and force-dependent channel transitions. Machine learning-based analysis improves detection of stochastic ion channel behavior, prediction of transition rates, and reconstruction of mechanotransduction dynamics. These computational approaches may facilitate multiscale mechanobiological modeling, AI-assisted drug discovery, and precision mechanotransduction research in cardiovascular disease.
Machine learning approaches are especially well-suited for mechanosensitive ion channel analysis because electrophysiological recordings often resemble time-series datasets containing transient stochastic events embedded within background noise. RCNN and LSTM architectures are capable of learning temporal dependencies and identifying recurring activation patterns associated with hidden channel states. These approaches can automatically classify gating events, estimate transition probabilities, and reconstruct likely kinetic schemes from raw patch-clamp recordings without requiring extensive manual intervention.
Several computational advantages arise from applying machine learning techniques to PIEZO channel analysis:
-
-
Improved identification of hidden gating states within noisy electrophysiological recordings
-
-
Faster and more robust analysis of large datasets
-
-
Enhanced prediction of transition rates and force-dependent kinetics
-
-
Reduced subjectivity compared with manual kinetic fitting
-
-
Potential integration with multiscale mechanobiological simulations
-
-
Improved ability to model nonlinear force-response relationships
Deep learning-based kinetic analysis has already demonstrated success in broader ion channel research. For example, Celik et al. [68] developed Deep-Channel, a neural-network-based framework capable of identifying single-molecule ion channel events from patch-clamp recordings with improved speed and accuracy relative to traditional methods. Similar approaches may prove highly valuable for PIEZO channels because mechanotransduction involves complex stochastic gating transitions that are difficult to resolve experimentally.
Computational modeling has also become increasingly important for understanding the structural mechanics underlying PIEZO activation. Molecular dynamics simulations, continuum membrane mechanics models, and multiscale biophysical simulations have provided insights into how membrane tension, curvature, and lipid interactions influence PIEZO conformational changes [69,70]. Structural simulations suggest that mechanical forces induce flattening of the curved membrane dome surrounding the channel, resulting in propagation of conformational changes toward the central pore region [50,71]. Computational studies further indicate that the energetic coupling between membrane deformation and channel gating plays a central role in mechanosensitivity.
Recent advances in cryo-electron microscopy and structural biology have also enabled integration of experimental and computational approaches to investigate PIEZO channel dynamics at near-atomic resolution [16,17]. Structural modeling combined with machine learning may eventually enable the prediction of channel responses to diverse mechanical stimuli across different physiological conditions. Such integrative frameworks could provide unprecedented insights into how PIEZO channels function within endothelial cells, cardiomyocytes, fibroblasts, and other mechanically active cardiovascular tissues.
Beyond mechanistic understanding, computational modeling of PIEZO channels holds significant translational potential. Accurate kinetic models may facilitate virtual drug screening, prediction of pharmacological responses, and identification of therapeutic targets for cardiovascular diseases involving abnormal mechanotransduction. Machine learning-assisted simulations could help identify compounds capable of modulating PIEZO gating behavior, membrane interactions, or downstream signaling pathways [72]. In cardiovascular medicine, such approaches may prove valuable for developing therapies targeting hypertension, cardiac hypertrophy, fibrosis, endothelial dysfunction, and ischemic injury.
Computational approaches may additionally support the development of personalized mechanobiological models that incorporate patient-specific biomechanical and genetic information. As precision medicine continues to expand, integrating electrophysiological data, molecular simulations, and artificial intelligence may allow individualized prediction of mechanotransduction responses and disease susceptibility.
Overall, advances in computational modeling and machine learning are rapidly transforming the study of PIEZO channel kinetics. By combining high-resolution electrophysiology, structural biology, biophysical simulations, and artificial intelligence, researchers are beginning to unravel the complex force-dependent dynamics governing mechanosensitive ion channel behavior. These emerging technologies are expected to play an increasingly important role in understanding cardiovascular mechanotransduction and in facilitating future therapeutic development targeting PIEZO-mediated signaling pathways.
3. Physiological Roles of PIEZO Channels in the Cardiovascular System
PIEZO channels are expressed in multiple cardiovascular cell types and regulate mechanosensitive signaling pathways involved in vascular homeostasis, myocardial adaptation, extracellular matrix remodeling, and blood cell deformability. Figure 3 summarizes the major cardiovascular cell populations and representative PIEZO-mediated signaling mechanisms discussed in the following sections. Figure 3A illustrates endothelial cell mechanotransduction in response to fluid shear stress generated by blood flow. Activation of endothelial PIEZO1 induces calcium influx and downstream signaling pathways involved in nitric oxide production, ATP release, endothelial alignment, vascular tone regulation, and maintenance of vascular homeostasis. Figure 3B depicts PIEZO signaling in cardiomyocytes during cyclic stretch and mechanical loading. Mechanical activation of PIEZO1 contributes to calcium homeostasis, excitation-contraction coupling, hypertrophic signaling, and mechano-chemo transduction pathways associated with cardiac adaptation and remodeling. Figure 3C demonstrates PIEZO-mediated mechanotransduction in cardiac fibroblasts, where mechanical stimulation activates inflammatory and profibrotic signaling pathways, including interleukin-6 secretion, p38 MAPK activation, fibroblast differentiation, and extracellular matrix remodeling. Figure 3D illustrates the role of PIEZO1 in erythrocyte mechanobiology. During passage through narrow microvasculature, red blood cell deformation activates PIEZO1-dependent mechanosensitive ion transport pathways that regulate intracellular calcium signaling, cell volume homeostasis, and erythrocyte deformability. Collectively, these cell-specific mechanotransduction mechanisms highlight the diverse and integrated roles of PIEZO channels in cardiovascular physiology and disease progression.
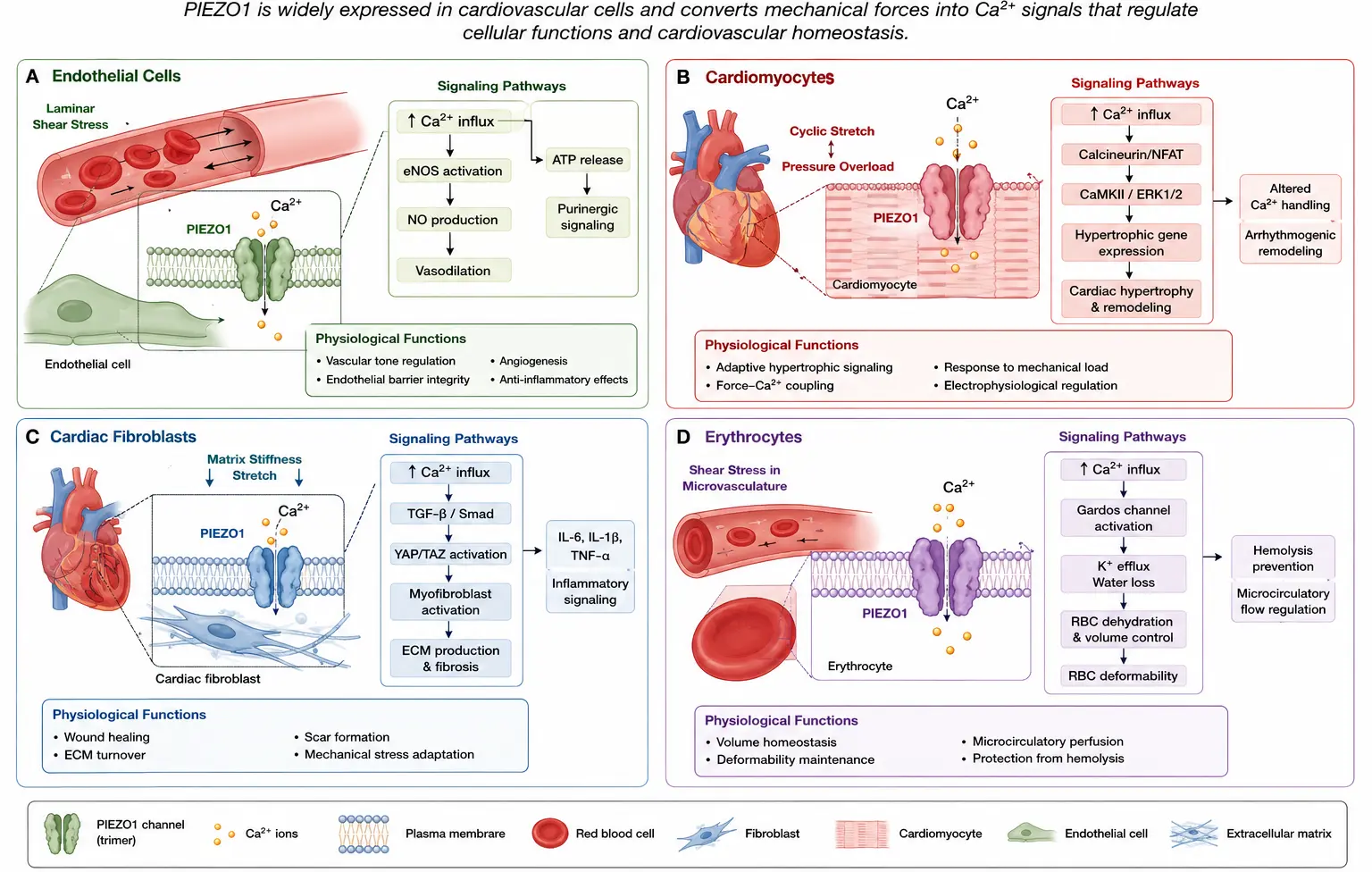
Figure 3. Cardiovascular cell-specific PIEZO signaling pathways. (A) Endothelial cell mechanotransduction mediated by PIEZO1 activation in response to fluid shear stress, resulting in calcium influx, nitric oxide production, ATP release, endothelial alignment, and vascular tone regulation. (B) Cardiomyocyte PIEZO signaling during mechanical loading and stretch, illustrating calcium-dependent pathways involved in excitation-contraction coupling, hypertrophic signaling, and mechano-chemo transduction. (C) Cardiac fibroblast mechanotransduction mediated by PIEZO1 activation, promoting inflammatory signaling, interleukin-6 secretion, p38 MAPK activation, fibroblast differentiation, and extracellular matrix remodeling. (D) Erythrocyte mechanobiology is regulated by PIEZO1-dependent mechanosensitive ion transport during microvascular deformation, contributing to red blood cell volume regulation, calcium signaling, and cellular deformability.
3.1. Endothelial Mechanotransduction
Endothelial cells form the inner lining of blood vessels and are continuously exposed to hemodynamic forces generated by circulating blood, including laminar shear stress, turbulent flow, hydrostatic pressure, and cyclic stretch [1,2]. These biomechanical forces are fundamental regulators of vascular physiology, influencing endothelial morphology, vascular tone, inflammatory signaling, permeability, angiogenesis, and vascular remodeling. To maintain cardiovascular homeostasis, endothelial cells must possess highly sensitive mechanotransduction systems capable of detecting subtle changes in blood flow and rapidly converting them into intracellular biochemical responses. Among the mechanosensitive signaling pathways identified in endothelial biology, PIEZO1 has emerged as one of the most important endothelial mechanosensors [26,29].
Endothelial mechanotransduction is coordinated through multiple interacting mechanosensory systems, including integrin-associated focal adhesions, cytoskeletal force transmission, extracellular matrix interactions, and mechanosensitive ion channels [5]. Integrin-mediated activation of focal adhesion kinase (FAK) and Src-family kinases contributes importantly to endothelial adaptation under laminar flow conditions by regulating cytoskeletal remodeling, nitric oxide signaling, and inflammatory responses. Emerging evidence suggests substantial crosstalk between integrin signaling pathways and PIEZO1-mediated calcium influx during vascular mechanotransduction.
PIEZO1 is abundantly expressed in vascular endothelial cells, where it directly senses fluid shear stress generated by blood flow [29]. Activation of PIEZO1 occurs when shear stress increases membrane tension and induces conformational changes within the channel structure, leading to calcium influx and downstream mechanotransduction signaling [18,50,60]. This calcium entry serves as a critical second messenger that regulates multiple endothelial functions, including nitric oxide production, ATP release, cytoskeletal remodeling, endothelial alignment, vascular tone regulation, and cell survival [27,30].
One of the most important physiological consequences of endothelial PIEZO1 activation is stimulation of nitric oxide signaling. Shear stress-induced calcium influx activates endothelial nitric oxide synthase (eNOS), leading to nitric oxide production and vasodilation [29,73]. Nitric oxide plays a central role in maintaining vascular homeostasis by reducing vascular resistance, inhibiting platelet aggregation, suppressing leukocyte adhesion, and limiting inflammatory activation. Through this mechanism, PIEZO1 contributes directly to the regulation of vascular tone and blood pressure.
In addition to regulating nitric oxide production, endothelial PIEZO1 signaling may interact with YAP/TAZ-mediated mechanotransduction pathways that respond dynamically to matrix stiffness and cytoskeletal tension [7]. Under disturbed or oscillatory flow conditions, altered mechanosensitive signaling may promote NF-κB activation, oxidative stress, endothelial inflammation, and maladaptive vascular remodeling associated with hypertension and atherosclerosis [5].
PIEZO1 also regulates endothelial alignment in response to directional blood flow. Under laminar flow conditions, endothelial cells elongate and align parallel to the direction of shear stress, thereby minimizing mechanical resistance and promoting vascular stability [1]. Li et al. [29] demonstrated that PIEZO1 is essential for this adaptive endothelial response. Inhibition or deletion of PIEZO1 disrupts flow-induced cellular alignment and impairs endothelial adaptation to hemodynamic forces, highlighting the critical role of PIEZO1 in vascular mechanosensing.
Another key endothelial function mediated by PIEZO1 is ATP release. Wang et al. [30] showed that activation of endothelial PIEZO1 triggers flow-induced ATP release into the extracellular environment. Extracellular ATP subsequently activates purinergic signaling pathways that contribute to vasodilation and cardiovascular regulation. Importantly, this mechanism was shown to play a significant role in blood pressure control. Endothelial-specific deletion of PIEZO1 impaired flow-mediated ATP release and altered systemic blood pressure regulation, demonstrating the physiological importance of PIEZO1-mediated mechanotransduction in vascular homeostasis.
The role of PIEZO1 extends beyond local endothelial signaling and contributes to whole-body cardiovascular adaptation. Rode et al. [74] demonstrated that PIEZO1 senses increases in blood flow associated with physical activity and exercise. During exercise, elevated shear stress activates endothelial PIEZO1, which contributes to resetting cardiovascular homeostasis and enhancing physical performance by promoting vascular adaptation and redistributing blood flow. These findings established PIEZO1 as an important sensor linking mechanical activity to systemic cardiovascular responses.
In addition to regulating adult vascular physiology, PIEZO1 is essential during embryonic vascular development. Ranade et al. [75] demonstrated that genetic deletion of PIEZO1 in mice results in severe vascular abnormalities and embryonic lethality. PIEZO1-deficient embryos exhibited defective vascular remodeling, impaired endothelial organization, and abnormal vessel architecture, indicating that endothelial mechanotransduction is indispensable for normal vascular morphogenesis. Similarly, Li et al. [29] showed that PIEZO1 integrates vascular architecture with physiological force sensing during vascular development. These studies collectively established that endothelial cells require PIEZO1-mediated mechanotransduction not only to maintain vascular homeostasis in adulthood but also to support proper cardiovascular development.
Recent investigations have further expanded the understanding of endothelial PIEZO1 signaling in vascular remodeling and inflammatory responses. Disturbed or oscillatory flow patterns, commonly observed at vascular bifurcations and regions prone to atherosclerosis, can alter PIEZO1 activation and promote endothelial inflammation [28,42]. Abnormal mechanotransduction under disturbed flow conditions contributes to endothelial dysfunction, leukocyte recruitment, oxidative stress, and vascular remodeling, thereby linking PIEZO1 to atherogenesis and the progression of vascular disease.
PIEZO1 has also been implicated in lymphatic endothelial mechanobiology. Choi et al. [43] demonstrated that PIEZO1 regulates flow-activated lymphatic expansion and lymphatic vessel remodeling through mechanotransduction-dependent pathways. These findings suggest that PIEZO-mediated endothelial signaling extends beyond the blood vasculature and contributes broadly to fluid homeostasis and vascular adaptation.
Mechanistically, endothelial PIEZO1 signaling is influenced by multiple factors, including membrane lipid composition, cytoskeletal organization, extracellular matrix stiffness, and local hemodynamic conditions [21,62]. Endothelial cells residing in stiffened or inflamed vascular environments may exhibit altered PIEZO1 sensitivity and abnormal calcium signaling, contributing to pathological vascular remodeling in hypertension and cardiovascular disease.
Chronic hemodynamic overload in hypertension alters endothelial mechanotransduction and may dysregulate PIEZO1-mediated calcium signaling pathways involved in vascular tone regulation, nitric oxide bioavailability, inflammatory activation, and vascular stiffness. Persistent endothelial dysfunction under abnormal flow conditions contributes to maladaptive vascular remodeling and progression of cardiovascular disease.
Overall, PIEZO1 functions as a master endothelial mechanosensor that enables blood vessels to continuously monitor and adapt to hemodynamic forces. Through regulation of calcium signaling, nitric oxide production, ATP release, endothelial alignment, and vascular remodeling, PIEZO1 plays a central role in cardiovascular physiology and vascular homeostasis. Dysregulation of endothelial PIEZO1 mechanotransduction contributes to hypertension, endothelial dysfunction, vascular inflammation, and atherosclerotic disease, highlighting its importance as both a physiological regulator and a potential therapeutic target in cardiovascular medicine.
3.2. Cardiac Mechanotransduction
The heart is a highly dynamic mechanical organ that continuously experiences complex biomechanical forces during each cardiac cycle, including stretch, compression, pressure overload, wall tension, and cyclic deformation [76,77]. Cardiomyocytes must constantly sense and adapt to these changing mechanical conditions in order to maintain efficient contractile performance and preserve cardiac homeostasis. This adaptive capability is mediated through sophisticated mechanotransduction pathways that convert mechanical stimuli into intracellular biochemical and electrophysiological responses [3]. Although integrins, cytoskeletal proteins, and stretch-activated channels have long been recognized as contributors to cardiac mechanosensing, recent evidence has established PIEZO1 as a major mechanosensitive ion channel involved in cardiac mechanotransduction [31,35].
PIEZO1 is expressed in cardiomyocytes and other cardiac cell types, where it functions as a mechanosensor responsive to membrane tension, stretch, and pressure overload [33,34]. Mechanical activation of PIEZO1 results in rapid calcium influx, triggering downstream signaling pathways involved in calcium homeostasis, excitation-contraction coupling, hypertrophic signaling, mechano-chemo transduction, and myocardial remodeling [31,39]. Because calcium signaling is central to cardiac physiology, PIEZO1-mediated mechanotransduction has profound effects on both acute cardiac function and long-term structural adaptation.
One of the most important functions of PIEZO1 in cardiomyocytes is the regulation of intracellular calcium homeostasis. Cardiomyocytes rely on tightly coordinated calcium cycling to control contraction and relaxation. Mechanical activation of PIEZO1 introduces an additional calcium entry pathway, linking mechanical loading directly to intracellular signaling networks [32]. Under physiological conditions, this mechanosensitive calcium influx may contribute to adaptive responses that optimize cardiac performance during exercise or transient increases in workload. However, excessive or sustained PIEZO1 activation under pathological conditions can disrupt calcium balance and promote maladaptive remodeling [39].
Recent studies have demonstrated that PIEZO1 participates directly in cardiac mechano-chemo transduction. Jiang et al. [31] showed that mechanical stimulation activates PIEZO1-dependent calcium signaling pathways in cardiomyocytes, thereby coupling mechanical stress to biochemical responses. Their findings established that PIEZO1 serves as an upstream mediator linking membrane deformation to intracellular signaling cascades involved in cardiac adaptation. This mechanosensitive signaling is especially important during changes in hemodynamic loading, where cardiomyocytes must rapidly adjust to altered pressure and stretch conditions.
PIEZO1 has also emerged as a critical regulator of pathological cardiac hypertrophy. Cardiac hypertrophy initially develops as a compensatory response to pressure overload, hypertension, or valvular disease, allowing the heart to maintain cardiac output under increased mechanical demand [76]. However, prolonged hypertrophic remodeling eventually becomes maladaptive and contributes to fibrosis, arrhythmias, and heart failure progression. Yu et al. [35] identified PIEZO1 as a key cardiac mechanosensor initiating pressure overload-induced hypertrophic signaling in adult mice. Their work demonstrated that mechanical stress activates PIEZO1 in cardiomyocytes, leading to calcium-dependent signaling pathways that promote hypertrophic growth and pathological remodeling.
Cardiac mechanotransduction involves coordinated signaling between mechanosensitive ion channels, integrins, focal adhesions, cytoskeletal proteins, extracellular matrix interactions, and transcriptional mechanoregulators [6]. Mechanical forces transmitted through focal adhesion complexes regulate MAPK signaling, calcium homeostasis, cytoskeletal remodeling, and hypertrophic gene expression. PIEZO1-mediated calcium influx likely functions synergistically with integrin-associated signaling pathways and stretch-sensitive TRP channels to regulate myocardial adaptation under physiological and pathological loading conditions.
Similarly, Liang et al. [33] reported that PIEZO1 expression is significantly upregulated in failing hearts and in cardiomyocytes stimulated with angiotensin II, suggesting an important role for PIEZO1 in cardiac stress responses. Wong et al. [34] further demonstrated that mechanical stretching activates PIEZO1-dependent signaling pathways associated with both physiological and pathological cardiac remodeling. These findings collectively indicate that PIEZO1 functions as a central mediator of cardiac adaptation to mechanical overload.
Mechanistically, PIEZO1-mediated calcium influx activates multiple downstream signaling pathways that regulate cardiac growth, remodeling, inflammation, and stress adaptation. One major pathway involves calpain activation. Calpains are calcium-dependent proteases that regulate cytoskeletal remodeling, protein turnover, and cellular stress responses. Excessive activation of calpain signaling contributes to cardiomyocyte dysfunction and structural remodeling during heart failure [39].
Another important downstream pathway involves calcineurin/NFAT signaling. Calcineurin is a calcium-sensitive phosphatase that activates nuclear factor of activated T-cells (NFAT) transcription factors, promoting expression of hypertrophic genes and pathological remodeling responses. Zhang et al. [39] demonstrated that PIEZO1-mediated calcium dysregulation activates calpain/calcineurin signaling, thereby promoting pressure overload-induced cardiac hypertrophy and fibrosis.
PIEZO1-mediated calcium influx additionally activates mitogen-activated protein kinase (MAPK) signaling pathways, including ERK1/2 and p38 MAPK, which regulate hypertrophic growth, inflammatory activation, and myocardial remodeling. Sustained activation of these pathways contributes to pathological cardiomyocyte hypertrophy, extracellular matrix remodeling, and progression toward heart failure under chronic pressure overload conditions.
PIEZO1 activation has additionally been linked to mitogen-activated protein kinase (MAPK) pathways, which regulate cellular growth, inflammatory signaling, stress responses, and myocardial remodeling [3]. MAPK activation contributes to cardiomyocyte hypertrophy and extracellular matrix remodeling during chronic mechanical stress. In parallel, calcium/calmodulin-dependent protein kinase II (CaMKII) signaling also plays a central role in cardiac mechanotransduction and in the progression of heart failure [78]. Because PIEZO1-mediated calcium influx can influence CaMKII activation, abnormal PIEZO signaling may contribute to arrhythmogenesis, contractile dysfunction, and maladaptive remodeling.
Mechanical stress also activates YAP/TAZ signaling pathways that regulate cardiomyocyte growth, fibrosis, and remodeling in response to altered matrix stiffness and cytoskeletal tension [7]. Increasing evidence suggests that PIEZO-mediated calcium signaling may interact with these mechanoregulated transcriptional pathways during cardiac hypertrophy and progression to heart failure.
Beyond cardiomyocytes, PIEZO-mediated mechanotransduction likely interacts with other components of the cardiac mechanosensory network, including integrins, stretch-sensitive cytoskeletal proteins, microtubules, and extracellular matrix signaling pathways [79]. Mechanical forces transmitted through the cytoskeleton can alter membrane tension and modulate PIEZO activation thresholds, further integrating mechanical and biochemical signaling within cardiac tissue.
Emerging evidence also suggests that PIEZO1 contributes to ischemic injury and adverse remodeling following myocardial infarction. Umbarkar et al. [40] demonstrated that mechanosensitive PIEZO1 activation aggravates ischemia-induced cardiac dysfunction and adverse remodeling. Excessive calcium influx through PIEZO1 during ischemic stress may promote oxidative injury, mitochondrial dysfunction, inflammatory activation, and cardiomyocyte death. These findings indicate that PIEZO1 may represent a mechanistic link between mechanical stress and ischemic pathophysiology.
Recent studies further suggest that altered PIEZO1 regulation may contribute to heart failure with preserved ejection fraction (HFpEF). Chen et al. [80] reported that trimethylamine N-oxide-induced cardiac diastolic dysfunction is associated with downregulation of PIEZO1 signaling, suggesting that both excessive and insufficient PIEZO activity may adversely affect cardiac function, depending on the disease context.
Collectively, current evidence establishes PIEZO1 as a central cardiac mechanosensor that links mechanical loading to intracellular calcium signaling and myocardial remodeling pathways. Through regulation of calcium homeostasis, mechano-chemo transduction, hypertrophic signaling, and excitation-contraction coupling, PIEZO1 enables cardiomyocytes to adapt to physiological mechanical stress while also contributing to pathological remodeling under chronic overload conditions. Continued investigation of PIEZO-mediated cardiac mechanotransduction may provide important insights into the mechanisms underlying cardiac hypertrophy, fibrosis, arrhythmias, and heart failure, while also identifying new therapeutic opportunities for cardiovascular disease.
3.3. Cardiac Fibroblasts and Matrix Remodeling
Cardiac fibroblasts are among the most abundant non-myocyte cell populations in the heart and play essential roles in maintaining structural integrity, extracellular matrix (ECM) turnover, tissue repair, and mechanical stability of the myocardium [36]. Under physiological conditions, fibroblasts regulate collagen synthesis and matrix homeostasis, thereby preserving normal myocardial architecture and mechanical function. However, following cardiac injury or chronic mechanical stress, fibroblasts become highly activated and contribute to pathological fibrosis, adverse remodeling, myocardial stiffening, and heart failure progression [3]. Because fibroblasts reside within a mechanically dynamic extracellular environment, they possess sophisticated mechanotransduction systems that sense changes in matrix stiffness, tissue deformation, and mechanical loading. Among these mechanosensitive pathways, PIEZO1 has emerged as an important regulator of fibroblast mechanobiology and cardiac remodeling [37,38].
Cardiac fibroblasts are highly mechanosensitive cells that respond rapidly to alterations in mechanical stress associated with myocardial infarction, hypertension, pressure overload, and ventricular dilation [36]. Mechanical activation of PIEZO1 in fibroblasts results in calcium influx and initiation of downstream signaling pathways involved in inflammation, fibroblast differentiation, ECM remodeling, and profibrotic responses [37]. These mechanotransduction pathways allow fibroblasts to adapt to changing mechanical environments but may also drive pathological fibrosis when chronically activated.
One of the key findings linking PIEZO1 to cardiac fibroblast biology was demonstrated by Blythe et al. [37], who showed that mechanical activation of PIEZO1 stimulates inflammatory signaling and cytokine production in cardiac fibroblasts. Specifically, PIEZO1 activation induced interleukin-6 (IL-6) secretion via calcium-dependent pathways that activated p38 mitogen-activated protein kinase (MAPK). IL-6 is a multifunctional pro-inflammatory cytokine implicated in cardiac hypertrophy, fibrosis, and heart failure progression. Elevated IL-6 signaling contributes to fibroblast activation, immune cell recruitment, and extracellular matrix deposition, thereby linking mechanotransduction directly to inflammatory cardiac remodeling.
PIEZO1-mediated fibroblast activation additionally interacts with transforming growth factor-β (TGF-β)/Smad signaling pathways, which are central regulators of cardiac fibrosis and extracellular matrix remodeling. Sustained activation of these mechanosensitive pathways promotes fibroblast differentiation, myofibroblast persistence, and progressive myocardial stiffening during chronic cardiovascular disease.
Activation of the p38 MAPK pathway represents another major consequence of fibroblast PIEZO1 signaling. MAPK pathways regulate cellular proliferation, differentiation, stress adaptation, and inflammatory responses under mechanical loading conditions [3]. Mechanical stimulation of fibroblast PIEZO1 promotes p38 MAPK activation, which subsequently enhances transcription of profibrotic and inflammatory genes [37]. Persistent activation of this pathway contributes to excessive collagen deposition and progressive myocardial stiffening during chronic cardiovascular disease.
PIEZO1 also plays a critical role in sensing extracellular matrix stiffness. Fibroblasts continuously monitor the biomechanical properties of their surrounding microenvironment, including matrix rigidity, tensile stress, and tissue deformation [38]. As fibrosis progresses and matrix stiffness increases, mechanical forces transmitted to fibroblasts become amplified, further enhancing mechanosensitive signaling pathways. Emig et al. [38] demonstrated that PIEZO1 contributes to stiffness sensing in human atrial fibroblasts and regulates cellular mechanical properties in response to altered matrix conditions. This creates a feed-forward cycle in which increased fibrosis leads to enhanced tissue stiffness, further activating PIEZO1-mediated profibrotic signaling and accelerating pathological remodeling.
Matrix stiffness sensing in fibroblasts is mediated through integrated mechanotransduction pathways involving integrins, focal adhesions, cytoskeletal tension, and YAP/TAZ transcriptional regulation [7]. Increased extracellular matrix rigidity amplifies mechanical force transmission to the plasma membrane, thereby enhancing PIEZO1-mediated calcium signaling and profibrotic activation. These interactions contribute to a feed-forward remodeling cycle in which fibrosis progressively increases tissue stiffness, further enhancing mechanosensitive signaling and pathological extracellular matrix deposition.
Mechanical activation of PIEZO1 additionally influences fibroblast differentiation into myofibroblasts, a specialized activated phenotype characterized by increased contractility, enhanced collagen synthesis, and elevated expression of alpha-smooth muscle actin (α-SMA) [36]. Myofibroblasts play central roles in scar formation and wound healing following myocardial injury; however, persistent myofibroblast activation contributes to maladaptive fibrosis and impaired cardiac compliance. Through calcium-dependent mechanotransduction pathways, PIEZO1 may regulate the differentiation and persistence of myofibroblasts under chronic mechanical stress conditions.
These mechanosensitive fibroblast responses are especially important during pathological cardiac remodeling following myocardial infarction. After ischemic injury, the infarcted myocardium undergoes substantial structural and mechanical changes characterized by inflammation, necrosis, scar formation, and altered wall stress distribution. Fibroblasts migrate into injured regions and become activated in response to inflammatory mediators and mechanical deformation [36]. PIEZO1-mediated mechanotransduction likely contributes to this reparative process by regulating cytokine production, matrix deposition, and fibroblast differentiation. However, excessive or prolonged activation may promote adverse ventricular remodeling, fibrosis, and progression to heart failure.
Similarly, chronic pressure overload resulting from hypertension or valvular disease exposes fibroblasts to sustained mechanical stress and increased matrix stiffness [76]. Under these conditions, persistent PIEZO1 activation may drive pathological extracellular matrix remodeling and myocardial stiffening. Fibrotic remodeling not only impairs cardiac relaxation and diastolic function but also disrupts electrical conduction pathways, increasing susceptibility to arrhythmias and heart failure.
Recent studies further suggest that PIEZO-mediated fibroblast signaling may interact with broader mechanobiological and epigenetic regulatory networks. Garoffolo and Pesce [81] highlighted emerging evidence linking cell mechanics and epitranscriptomic regulation in cardiac fibrosis, suggesting that mechanosensitive pathways involving PIEZO channels may influence RNA modification and transcriptional regulation during fibrotic remodeling. These findings point toward increasingly complex interactions between biomechanical signaling and gene regulation in cardiovascular disease.
PIEZO1-mediated fibroblast mechanotransduction is also influenced by extracellular matrix composition, cytoskeletal organization, and membrane mechanics. Changes in collagen crosslinking, matrix rigidity, and cytoskeletal tension alter force transmission to the plasma membrane and may modulate PIEZO activation thresholds [62]. Such interactions emphasize the importance of the biomechanical microenvironment in regulating fibroblast behavior and cardiac remodeling dynamics.
Overall, PIEZO1 functions as a critical mechanosensor in cardiac fibroblasts, linking mechanical stress to inflammatory signaling, matrix remodeling, and fibrosis. Through regulation of IL-6 secretion, p38 MAPK activation, matrix stiffness sensing, and fibroblast differentiation, PIEZO1 contributes to both adaptive tissue repair and pathological remodeling in the injured heart. Dysregulated fibroblast mechanotransduction plays a major role in myocardial fibrosis, ventricular stiffening, arrhythmogenesis, and progression of heart failure, highlighting PIEZO1 as a potentially important therapeutic target to limit adverse cardiac remodeling and fibrotic disease.
3.4. Red Blood Cell Mechanobiology
Red blood cells (RBCs), or erythrocytes, are continuously exposed to substantial mechanical stress as they circulate through the cardiovascular system. To efficiently transport oxygen and carbon dioxide, erythrocytes must repeatedly deform while traversing narrow capillaries, splenic sinusoids, and microvascular networks that are often smaller than the resting diameter of the cell itself [82]. This extraordinary deformability is essential for maintaining adequate tissue perfusion and microcirculatory flow. Consequently, erythrocytes require highly specialized mechanobiological mechanisms that can sense and adapt to mechanical deformation while preserving membrane integrity and cellular volume homeostasis. Among these mechanisms, PIEZO1 has emerged as a central mechanosensitive ion channel regulating erythrocyte physiology and biomechanical adaptation [26,83].
Unlike many other cell types, mature erythrocytes lack nuclei and most intracellular organelles, limiting their ability to respond to environmental stress through transcriptional regulation. Instead, they rely heavily on membrane-based mechanotransduction systems to rapidly sense and adapt to mechanical forces encountered during circulation. PIEZO1 functions as a mechanosensitive cation channel in the erythrocyte membrane, where it is activated by membrane stretching, compression, and deformation during microvascular passage [83]. Activation of PIEZO1 allows transient influx of cations, particularly calcium ions, thereby initiating downstream mechanisms involved in cell volume regulation and membrane adaptation.
Mechanical activation of PIEZO1 plays a major role in erythrocyte deformability and osmotic balance. During passage through narrow capillaries, RBC deformation transiently activates PIEZO1-mediated calcium influx, which subsequently stimulates ion transport pathways that regulate intracellular potassium and water content [83]. These transient calcium-mediated responses allow erythrocytes to dynamically adjust their hydration state and membrane flexibility during circulation. Proper regulation of cell volume is essential because excessive swelling or dehydration can impair deformability, increase blood viscosity, and compromise microvascular perfusion.
Danielczok et al. [83] demonstrated that red blood cell passage through small capillaries is associated with transient calcium-mediated adaptive responses involving PIEZO1 activation. These findings established a direct mechanistic link between mechanical deformation and the regulation of ion transport in erythrocytes. PIEZO1-mediated mechanotransduction, therefore, enables RBCs to function as highly responsive biomechanical systems that adapt to constantly changing hemodynamic environments.
The importance of PIEZO1 in erythrocyte physiology is further illustrated by hereditary disorders associated with PIEZO1 mutations. Gain-of-function mutations in PIEZO1 are strongly linked to hereditary xerocytosis, a rare hemolytic anemia characterized by erythrocyte dehydration, impaired deformability, and chronic hemolysis [13,56]. In hereditary xerocytosis, abnormal PIEZO1 activation leads to excessive cation leakage, altered potassium transport, and chronic cellular dehydration. These changes reduce erythrocyte flexibility and increase susceptibility to mechanical damage during circulation, ultimately contributing to premature RBC destruction.
The pathophysiology of hereditary xerocytosis highlights the critical role of mechanosensitive ion transport in maintaining erythrocyte homeostasis. Even subtle alterations in PIEZO1 gating kinetics or mechanosensitivity can disrupt intracellular ionic balance and profoundly affect red blood cell survival and mechanical performance. These findings also emphasize that PIEZO1 must maintain tightly regulated activation and inactivation kinetics to prevent excessive calcium loading and membrane dysfunction.
In addition to hereditary xerocytosis, altered erythrocyte mechanobiology may contribute to broader cardiovascular and hematological disorders. Impaired RBC deformability is associated with microvascular dysfunction, thrombosis, inflammation, and tissue hypoxia in conditions such as diabetes mellitus, sickle cell disease, hypertension, and sepsis [82]. Because PIEZO1 directly regulates erythrocyte mechanical adaptation, abnormal PIEZO signaling may influence blood rheology and microcirculatory function under pathological conditions.
Recent studies further suggest that PIEZO1-mediated mechanotransduction may interact with endothelial and vascular signaling pathways. Mechanically activated erythrocytes can release ATP and other signaling molecules that influence vascular tone and endothelial responses, potentially contributing to microvascular regulation and tissue perfusion [2,30]. Thus, erythrocyte PIEZO1 signaling may participate not only in cellular adaptation but also in broader cardiovascular communication networks.
Mechanistically, erythrocyte PIEZO1 activity is influenced by membrane lipid composition, cytoskeletal organization, and mechanical loading conditions. The erythrocyte membrane possesses a highly specialized spectrin-actin cytoskeletal network that provides both flexibility and mechanical resilience. Interactions between PIEZO1 and the membrane cytoskeleton likely influence mechanosensitivity, force transmission, and channel gating behavior during repeated deformation cycles [21,62]. Because erythrocytes undergo millions of deformation events during their lifespan, maintaining stable mechanotransduction mechanisms is essential for long-term cellular survival.
Emerging biophysical studies have also begun to investigate the kinetics and permeation properties of PIEZO channels in erythrocytes at the molecular level [57]. Understanding these gating dynamics may help clarify how PIEZO1 balances mechanosensitivity with cellular protection under continuous hemodynamic stress. Recent work describing spring-like mechanical gating behavior and rapid inactivation kinetics may be particularly relevant to erythrocytes, where transient mechanosensitive responses are required to prevent sustained ionic imbalance [53].
Overall, PIEZO1 plays a central role in red blood cell mechanobiology by regulating mechanosensitive ion transport, cellular deformability, and volume homeostasis. Through its ability to sense membrane deformation and mediate adaptive calcium signaling, PIEZO1 enables erythrocytes to survive the extreme mechanical challenges of microvascular circulation. Mutations that disrupt PIEZO1 function result in severe erythrocyte abnormalities, such as hereditary xerocytosis, underscoring the physiological importance of mechanotransduction in RBC homeostasis. Continued investigation of erythrocyte PIEZO signaling may provide important insights into microvascular physiology, blood rheology, and mechanisms of cardiovascular disease.
Table 1 summarizes the major cardiovascular and mechanically active cell types in which PIEZO-mediated mechanotransduction plays important physiological and pathological roles. The table highlights the diverse mechanical stimuli sensed by PIEZO channels, including fluid shear stress, cyclic stretch, pressure overload, membrane deformation, and extracellular matrix stiffness, as well as the principal physiological functions and downstream signaling pathways activated in different cellular environments. In endothelial cells, PIEZO1 primarily functions as a shear stress sensor, regulating nitric oxide production, ATP release, endothelial alignment, vascular tone, and vascular remodeling. In cardiomyocytes, PIEZO-mediated calcium signaling contributes to excitation-contraction coupling, mechano-chemo transduction, hypertrophic remodeling, and adaptive responses to mechanical loading. Cardiac fibroblasts utilize PIEZO1 to detect matrix stiffness and tissue deformation, thereby promoting inflammatory signaling, fibroblast differentiation, extracellular matrix remodeling, and fibrosis. In erythrocytes, PIEZO1 regulates mechanosensitive ion transport, cellular deformability, osmotic balance, and volume homeostasis during microvascular circulation. Additional mechanically responsive cell populations, including vascular smooth muscle cells, monocytes, and lymphatic endothelial cells, further illustrate the broad physiological importance of PIEZO-mediated mechanotransduction throughout the cardiovascular system. Collectively, these integrated mechanobiological pathways demonstrate how PIEZO channels couple biomechanical forces to cellular signaling, tissue remodeling, inflammation, and cardiovascular disease progression across multiple biological scales.
Table 1. Cell-specific roles of PIEZO-mediated mechanotransduction in the cardiovascular system.
|
Cell Type |
Mechanical Stimulus |
PIEZO Function |
Physiological Function |
Key Signaling Pathways |
Disease Relevance |
Representative References |
|---|---|---|---|---|---|---|
|
Endothelial cells |
Fluid shear stress, laminar flow, disturbed flow, cyclic stretch |
Shear stress sensing and endothelial mechanotransduction |
Regulation of vascular tone, nitric oxide production, ATP release, endothelial alignment, vascular remodeling, blood pressure homeostasis |
Ca2+ influx, eNOS activation, ATP–purinergic signaling, MAPK signaling, inflammatory signaling |
Hypertension, endothelial dysfunction, atherosclerosis, vascular inflammation, vascular remodeling |
|
|
Cardiomyocytes |
Cyclic stretch, pressure overload, wall tension, mechanical deformation |
Cardiac mechanosensing and mechano-chemo transduction |
Regulation of calcium homeostasis, excitation-contraction coupling, adaptive remodeling, stress sensing |
Ca2+ signaling, calpain activation, calcineurin/NFAT signaling, MAPK pathways, CaMKII activation |
Cardiac hypertrophy, arrhythmias, heart failure, ischemic injury, maladaptive remodeling |
|
|
Cardiac fibroblasts |
Matrix stiffness, tensile stress, tissue deformation, mechanical overload |
Matrix mechanosensing and profibrotic mechanotransduction |
Extracellular matrix remodeling, fibroblast activation, cytokine secretion, wound healing, scar formation |
Ca2+ influx, IL-6 signaling, p38 MAPK activation, TGF-β signaling, YAP/TAZ activation |
Cardiac fibrosis, ventricular stiffening, adverse remodeling, arrhythmogenesis |
|
|
Erythrocytes (RBCs) |
Membrane deformation, capillary compression, shear stress in microcirculation |
Mechanosensitive ion transport and deformation sensing |
Regulation of cell volume, deformability, osmotic balance, microvascular adaptation |
Ca2+ influx, Gardos channel activation, potassium efflux, water transport regulation |
Hereditary xerocytosis, hemolysis, impaired microcirculation, altered blood rheology |
|
|
Vascular smooth muscle cells |
Circumferential stretch, pressure overload, vessel wall tension |
Vascular mechanosensing and contractile regulation |
Regulation of vascular contraction, vessel stiffness, adaptive remodeling |
Ca2+ signaling, RhoA/ROCK signaling, MAPK activation |
Hypertension, vascular stiffening, arterial remodeling |
|
|
Monocytes/immune cells |
Disturbed shear stress, mechanical deformation during circulation |
Inflammatory mechanotransduction |
Cytokine release, endothelial interaction, inflammatory activation |
NF-κB signaling, inflammatory cytokine signaling, Ca2+-dependent activation |
Atherosclerosis, vascular inflammation, endothelial injury |
[42] |
|
Lymphatic endothelial cells |
Interstitial flow, lymphatic shear stress, vessel stretch |
Flow sensing and lymphatic mechanotransduction |
Lymphatic remodeling, lymphatic expansion, fluid homeostasis |
Ca2+ signaling, mechanosensitive endothelial pathways |
Lymphatic dysfunction, impaired fluid homeostasis |
[86] |
4. PIEZO Channels in Cardiovascular Disease
Impaired mechanotransduction is increasingly recognized as a major contributor to the progression of cardiovascular disease. In addition to PIEZO channels, abnormal integrin signaling, focal adhesion remodeling, cytoskeletal dysfunction, extracellular matrix stiffening, and altered YAP/TAZ activity contribute importantly to endothelial dysfunction, vascular inflammation, cardiac hypertrophy, and fibrosis [5,6]. PIEZO-mediated calcium influx may interact with these broader mechanosensory pathways to regulate inflammatory activation, oxidative stress, extracellular matrix remodeling, and maladaptive tissue adaptation under chronic biomechanical overload.
Defective PIEZO-mediated mechanotransduction contributes to multiple forms of cardiovascular pathology by linking abnormal mechanical stress to inflammatory signaling, calcium dysregulation, fibrosis, vascular dysfunction, and adverse remodeling. Figure 4 summarizes the major cardiovascular diseases and pathological remodeling processes associated with dysregulated PIEZO-mediated mechanotransduction. Mechanical stress, altered hemodynamic forces, and abnormal membrane tension activate PIEZO-dependent calcium signaling pathways that contribute to multiple forms of cardiovascular pathology. Figure 4A illustrates the role of PIEZO signaling in hypertension, where abnormal endothelial mechanotransduction promotes endothelial dysfunction, vascular stiffening, inflammatory activation, and impaired vascular tone regulation. Figure 4B depicts PIEZO-mediated cardiac fibrosis and extracellular matrix remodeling, highlighting fibroblast activation, inflammatory cytokine production, myofibroblast differentiation, and excessive collagen deposition that contribute to myocardial stiffening and adverse remodeling. Figure 4C demonstrates the involvement of PIEZO channels in cardiac hypertrophy and heart failure progression through calcium overload and activation of downstream hypertrophic signaling pathways, including calpain/calcineurin, MAPK, and CaMKII signaling. Figure 4D illustrates the contribution of PIEZO-mediated mechanotransduction to vascular inflammation and atherosclerosis, in which disturbed flow activates endothelial cells and circulating monocytes, thereby promoting inflammatory signaling and vascular injury. Figure 4E summarizes the role of PIEZO signaling during ischemic injury and reperfusion, including oxidative stress, mitochondrial dysfunction, inflammatory activation, and cardiomyocyte death that contribute to adverse cardiac remodeling. Collectively, these interconnected mechanisms demonstrate how abnormal mechanotransduction can drive cardiovascular disease progression across multiple biological scales.
4.1. Hypertension
PIEZO1 is highly expressed in vascular endothelial cells, where it directly senses fluid shear stress generated by blood flow [29]. Under physiological conditions, shear stress-induced activation of PIEZO1 promotes calcium influx and downstream signaling pathways that regulate nitric oxide production, ATP release, endothelial alignment, and vasodilation [30]. These adaptive responses allow blood vessels to maintain appropriate vascular resistance and tissue perfusion despite changing hemodynamic demands. Through its role in endothelial mechanotransduction, PIEZO1 therefore functions as an important determinant of systemic blood pressure homeostasis.
Disruption of normal PIEZO1 signaling can profoundly impair vascular adaptation and contribute to the development of hypertension. Dysregulated mechanotransduction may alter endothelial responsiveness to shear stress, impair vasodilatory signaling, and promote pathological vascular remodeling [26]. One major consequence of abnormal PIEZO1 activity is endothelial dysfunction, a hallmark feature of hypertension and vascular disease. Endothelial dysfunction is characterized by reduced nitric oxide bioavailability, impaired vasodilation, increased oxidative stress, and heightened inflammatory activation. Because PIEZO1-mediated calcium signaling directly activates endothelial nitric oxide synthase, altered PIEZO1 function may compromise endothelium-dependent vasorelaxation and promote elevated vascular resistance [30].
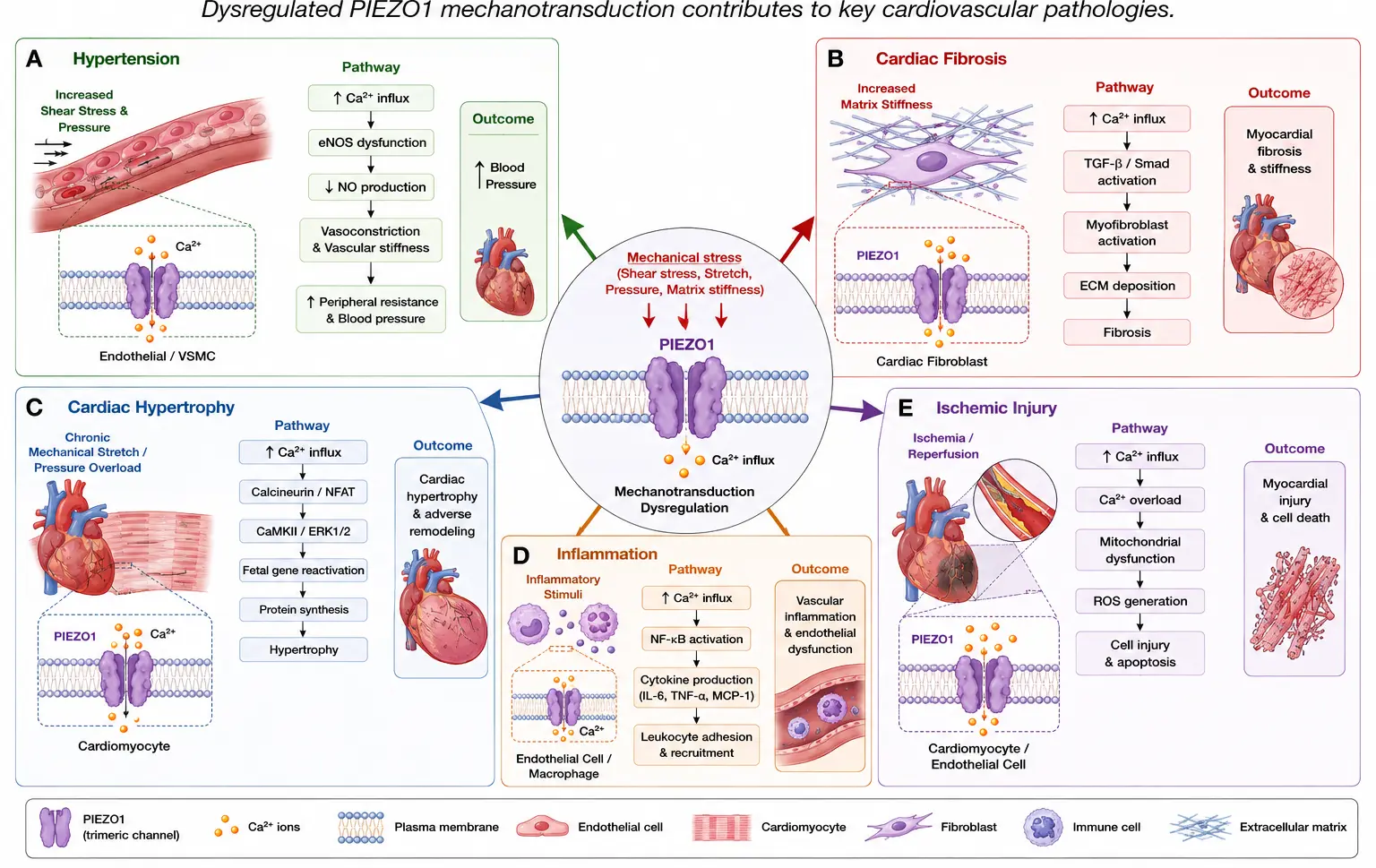
Figure 4. Roles of PIEZO-mediated mechanotransduction in cardiovascular disease. Schematic overview illustrating the involvement of PIEZO channels in major cardiovascular pathologies. Mechanical stress, altered hemodynamics, and abnormal membrane tension activate PIEZO-mediated calcium signaling pathways that contribute to (A) hypertension through endothelial dysfunction, vascular stiffening, and abnormal vasoconstriction; (B) cardiac fibrosis through fibroblast activation, inflammatory signaling, and extracellular matrix remodeling; (C) cardiac hypertrophy through calcium dysregulation and activation of hypertrophic signaling pathways including calpain/calcineurin and MAPK signaling; (D) vascular inflammation and atherosclerosis through endothelial activation, monocyte mechanotransduction, and inflammatory cytokine production; and (E) ischemic injury and adverse cardiac remodeling through oxidative stress, mitochondrial dysfunction, inflammatory activation, and cardiomyocyte death. Collectively, these mechanisms demonstrate the central role of PIEZO-mediated mechanotransduction in cardiovascular disease progression.
Abnormal PIEZO signaling also contributes to vascular stiffening, another key pathological feature of hypertension. Chronic mechanical stress and elevated blood pressure progressively alter vascular wall structure through extracellular matrix remodeling, collagen deposition, and smooth muscle hypertrophy [2]. Increased vascular stiffness amplifies pulse wave propagation and further elevates systolic blood pressure, creating a vicious cycle of mechanical overload and vascular injury. Because PIEZO1 functions as a mechanosensor responsive to membrane tension and mechanical stress, persistent hypertension may chronically activate PIEZO1-Mediated signaling pathways, accelerating vascular remodeling and stiffening [27].
In addition to structural remodeling, dysregulated PIEZO1 activity may promote abnormal vasoconstriction. Under physiological conditions, endothelial PIEZO1 contributes to balanced regulation of vascular tone through nitric oxide and purinergic signaling pathways [30]. However, impaired mechanotransduction may shift the balance toward enhanced vasoconstrictive signaling, increasing peripheral vascular resistance and sustaining hypertension. Altered calcium handling associated with abnormal PIEZO activity may further disrupt vascular smooth muscle contractility and endothelial communication.
Enhanced inflammatory signaling represents another important mechanism linking PIEZO dysregulation to hypertension. Increasing evidence indicates that chronic vascular inflammation contributes substantially to endothelial dysfunction, arterial remodeling, and hypertensive organ damage [42]. Mechanical stress-induced activation of PIEZO1 can stimulate inflammatory pathways involving cytokine release, oxidative stress, and leukocyte recruitment. Disturbed flow patterns and abnormal shear stress conditions, commonly present in hypertensive vasculature, may exacerbate PIEZO-mediated inflammatory signaling and vascular injury [73]. Such inflammatory responses contribute not only to vascular dysfunction but also to progressive remodeling of the heart, kidneys, and microvasculature.
Recent studies have highlighted the potential role of altered PIEZO1 signaling in hypertensive nephropathy and renal vascular pathology. Nagase and Nagase [41] proposed that PIEZO ion channels function as long-sought-after mechanosensors that mediate hypertension-associated vascular and renal injury. In the kidney, elevated blood pressure generates substantial mechanical stress within glomerular and vascular structures, potentially activating PIEZO-dependent signaling pathways that contribute to endothelial injury, fibrosis, and progressive nephropathy. Chronic activation of mechanosensitive pathways may therefore represent an important link between hypertension and end-organ damage.
Emerging evidence also suggests that PIEZO-mediated mechanotransduction may interact with broader neurohumoral and cardiovascular regulatory systems involved in blood pressure control. Mechanosensitive signaling influences autonomic regulation, vascular reactivity, renal sodium handling, and inflammatory responses, all of which contribute to long-term blood pressure homeostasis [87]. Because PIEZO1 integrates mechanical stimuli with calcium-dependent intracellular signaling, it may serve as a central node connecting hemodynamic stress to systemic cardiovascular regulation.
At the molecular level, PIEZO1 activity in hypertension is likely influenced by multiple factors, including membrane stiffness, extracellular matrix composition, cytoskeletal organization, oxidative stress, and lipid membrane properties [21,62]. Hypertensive vascular remodeling alters the biomechanical environment surrounding endothelial cells, potentially modifying PIEZO activation thresholds and mechanosensitivity. Increased extracellular matrix stiffness may amplify force transmission to endothelial membranes, thereby altering mechanotransduction dynamics and perpetuating vascular dysfunction.
Recent pharmacological studies have further emphasized the therapeutic relevance of PIEZO channels in hypertension. Because PIEZO1 directly participates in blood pressure regulation and vascular remodeling, modulation of PIEZO1-Mediated signaling may represent a novel therapeutic strategy for treating hypertension and associated vascular diseases [72]. However, given the widespread physiological functions of PIEZO1 throughout the cardiovascular system, careful targeting strategies will be necessary to avoid adverse effects on normal mechanosensory function.
Overall, current evidence strongly supports a major role for PIEZO1-mediated mechanotransduction in the regulation of vascular homeostasis and blood pressure control. Dysregulated PIEZO signaling contributes to endothelial dysfunction, vascular stiffening, abnormal vasoconstriction, inflammatory activation, and hypertensive organ injury. Continued investigation of PIEZO-dependent vascular signaling pathways may provide important insights into the mechanisms underlying hypertension and identify new therapeutic opportunities for preventing cardiovascular and renal complications associated with chronic elevated blood pressure.
4.2. Cardiac Hypertrophy and Heart Failure
Because mechanical stress is one of the primary triggers of cardiac hypertrophy, mechanotransduction pathways play central roles in regulating the transition from adaptive remodeling to pathological heart failure. Among the mechanosensitive pathways implicated in this process, PIEZO1 has emerged as a key cardiac mechanosensor linking mechanical overload to maladaptive intracellular signaling [33,35].
Cardiomyocytes are continuously exposed to dynamic biomechanical forces during each cardiac cycle, including stretch, pressure, wall tension, and cyclic deformation. Under pathological conditions such as chronic hypertension or aortic stenosis, sustained mechanical overload dramatically increases myocardial stress and activates mechanosensitive signaling pathways [76]. PIEZO1, as a mechanically activated cation channel, responds directly to these forces by mediating calcium influx into cardiomyocytes [31]. Because intracellular calcium is a central regulator of cardiac contraction, metabolism, and gene transcription, abnormal PIEZO1 activation can profoundly influence cardiac remodeling and disease progression.
Evidence linking PIEZO1 to pathological cardiac hypertrophy has increased substantially in recent years. Liang et al. [33] demonstrated that PIEZO1 expression is significantly upregulated in failing hearts as well as in cardiomyocytes stimulated with angiotensin II (AngII), a major neurohormonal mediator of hypertrophic remodeling. AngII-induced mechanical and biochemical stress increased PIEZO1 expression and mechanosensitive signaling, suggesting that PIEZO1 participates in the pathological response to chronic cardiac overload. These findings provided early evidence that PIEZO1 activation may contribute directly to maladaptive cardiac remodeling during heart failure progression.
Subsequent studies further clarified the molecular mechanisms underlying PIEZO1-mediated hypertrophic signaling. Zhang et al. [39] demonstrated that mechanical activation of PIEZO1 disrupts intracellular calcium homeostasis and activates calpain/calcineurin signaling pathways, thereby promoting pathological hypertrophy. In their study, PIEZO1-mediated calcium influx stimulated calcium-dependent proteases known as calpains, which regulate cytoskeletal remodeling, protein degradation, and stress responses within cardiomyocytes. Excessive calpain activation contributes to structural disorganization, contractile dysfunction, and myocardial injury during chronic mechanical overload.
PIEZO1-mediated calcium dysregulation also activates calcineurin, a calcium-sensitive phosphatase that dephosphorylates nuclear factor of activated T-cells (NFAT) transcription factors. Activated NFAT translocates into the nucleus and induces expression of hypertrophic and profibrotic genes, thereby driving pathological myocardial remodeling [39]. This calcineurin/NFAT pathway is widely recognized as one of the central signaling mechanisms underlying maladaptive cardiac hypertrophy and heart failure progression.
In addition to calpain and calcineurin signaling, PIEZO1 activation influences several other downstream pathways involved in cardiac remodeling. Mechanosensitive calcium influx can stimulate mitogen-activated protein kinase (MAPK) pathways, which regulate cellular growth, inflammatory responses, oxidative stress, and fibrosis [3]. PIEZO1-mediated signaling may also contribute to activation of calcium/calmodulin-dependent protein kinase II (CaMKII), a critical regulator of cardiac excitation-contraction coupling, arrhythmogenesis, and heart failure progression [78]. Persistent CaMKII activation has been strongly linked to electrical instability, impaired calcium cycling, and ventricular dysfunction in failing hearts.
The direct role of PIEZO1 in pressure overload-induced remodeling was further demonstrated by Yu et al. [35], who identified PIEZO1 as a key cardiac mechanosensor initiating hypertrophic responses in adult mice. Importantly, genetic deletion of PIEZO1 significantly attenuated pressure overload-induced cardiac remodeling, fibrosis, and ventricular dysfunction. These findings established that PIEZO1 is not merely associated with hypertrophic remodeling but actively contributes to disease progression through mechanotransduction-dependent signaling pathways.
Excessive or sustained PIEZO1 activation may therefore contribute to several pathological processes characteristic of heart failure:
-
-
Calcium overload and impaired calcium homeostasis
-
-
Activation of hypertrophic gene expression
-
-
Fibrotic remodeling and extracellular matrix deposition
-
-
Contractile dysfunction
-
-
Oxidative stress and mitochondrial injury
-
-
Electrical instability and arrhythmogenesis
-
-
Progressive ventricular remodeling and heart failure
Calcium overload is particularly important in the pathogenesis of heart failure because chronic intracellular calcium dysregulation impairs excitation-contraction coupling and promotes cellular injury. Under physiological conditions, transient calcium signaling is tightly regulated to coordinate myocardial contraction and relaxation. However, excessive PIEZO1 activation during chronic mechanical stress may lead to persistent calcium influx, mitochondrial dysfunction, oxidative stress, and activation of cell death pathways [39,40].
Fibrosis represents another major consequence of PIEZO-mediated cardiac remodeling. Mechanical overload stimulates fibroblast activation and extracellular matrix deposition, increasing myocardial stiffness and impairing diastolic relaxation [36]. Because PIEZO1 is expressed in both cardiomyocytes and fibroblasts, abnormal mechanotransduction may simultaneously promote hypertrophic signaling in myocytes and profibrotic remodeling in fibroblasts, thereby accelerating heart failure progression.
Recent evidence further suggests that PIEZO1 contributes to ischemic remodeling and post-infarction heart failure. Umbarkar et al. [40] demonstrated that mechanosensitive PIEZO1 activation aggravates ischemia-induced adverse cardiac remodeling and dysfunction. During ischemic injury, increased wall stress and altered tissue mechanics may excessively activate PIEZO1, leading to calcium overload, inflammatory signaling, and myocardial damage. These findings indicate that PIEZO1-mediated mechanotransduction contributes not only to chronic hypertensive remodeling but also to acute ischemic pathophysiology.
Emerging studies also suggest that altered PIEZO signaling may be involved in heart failure with preserved ejection fraction (HFpEF), a condition strongly associated with hypertension, fibrosis, and increased ventricular stiffness. Chen et al. [80] reported that trimethylamine N-oxide-induced diastolic dysfunction is associated with downregulation of PIEZO1 signaling, suggesting that both excessive activation and impaired mechanotransduction may adversely affect cardiac function depending on the disease context. These findings highlight the complex and context-dependent role of PIEZO1 in cardiac physiology and pathology.
Mechanistically, the effects of PIEZO1 on cardiac remodeling are influenced by the surrounding biomechanical environment, including extracellular matrix stiffness, cytoskeletal organization, membrane tension, and neurohumoral signaling pathways [79]. Progressive myocardial stiffening associated with fibrosis and hypertrophy may further amplify mechanosensitive signaling, creating a feed-forward cycle of mechanical stress and pathological remodeling.
Overall, accumulating evidence strongly supports a central role for PIEZO1-mediated mechanotransduction in cardiac hypertrophy and in the progression of heart failure. By linking mechanical overload to calcium-dependent signaling pathways, PIEZO1 regulates hypertrophic growth, fibrosis, contractile dysfunction, and maladaptive remodeling. Continued investigation of PIEZO-dependent cardiac mechanotransduction may provide important insights into the mechanisms underlying heart failure and identify new therapeutic strategies to limit pathological remodeling and preserve cardiac function.
4.3. Fibrosis and Extracellular Matrix Remodeling
Increasing evidence indicates that mechanotransduction pathways play critical roles in fibrosis and extracellular matrix (ECM) remodeling processes by linking altered tissue mechanics to profibrotic cellular responses. Among the mechanosensitive regulators implicated in cardiovascular fibrosis, PIEZO1 has emerged as an important mediator of fibroblast activation, inflammatory signaling, and matrix remodeling [37,38].
A defining characteristic of fibrosis is the progressive increase in tissue stiffness caused by excessive extracellular matrix accumulation. Mechanical stiffening itself profoundly alters the biomechanical microenvironment surrounding cardiovascular cells and amplifies mechanotransduction signaling pathways [2]. This creates a self-perpetuating feedback loop in which fibrosis increases matrix rigidity, enhanced stiffness activates mechanosensitive pathways such as PIEZO1, and further mechanotransduction signaling promotes additional fibrosis and remodeling. Consequently, mechanical stiffening and fibrosis form a self-amplifying cycle that contributes to progressive cardiovascular dysfunction.
PIEZO1-mediated mechanotransduction plays a major role in sensing these biomechanical alterations. In cardiac fibroblasts, mechanical stress activates PIEZO1 and induces calcium influx, which subsequently stimulates downstream inflammatory and profibrotic signaling pathways [37]. PIEZO1 activation promotes fibroblast differentiation, extracellular matrix synthesis, cytokine production, and matrix stiffness sensing, all of which contribute to fibrotic remodeling. Because fibroblasts are highly responsive to mechanical cues, altered tissue stiffness substantially influences their activation state and profibrotic behavior.
One of the major downstream consequences of PIEZO1 activation is stimulation of inflammatory signaling pathways. Blythe et al. [37] demonstrated that mechanically activated PIEZO1 channels in cardiac fibroblasts induce interleukin-6 (IL-6) secretion through p38 MAPK-dependent mechanisms. IL-6 is a potent inflammatory cytokine implicated in myocardial fibrosis, hypertrophy, and the progression of heart failure. Persistent inflammatory activation enhances extracellular matrix deposition and promotes the recruitment of immune cells, which further amplify fibrotic signaling. Thus, PIEZO1-mediated mechanotransduction provides an important link between mechanical stress and inflammatory remodeling within the cardiovascular system.
Mechanical activation of PIEZO1 also influences fibroblast differentiation into myofibroblasts, specialized contractile cells that produce large amounts of collagen and extracellular matrix proteins [36]. Myofibroblast activation is essential for wound healing and scar formation following myocardial injury; however, persistent activation contributes to pathological fibrosis, ventricular stiffening, and impaired cardiac relaxation. Through calcium-dependent signaling pathways, PIEZO1 likely regulates the persistence and activity of myofibroblasts under chronic mechanical stress conditions.
Extracellular matrix remodeling mediated by PIEZO1 has important consequences for myocardial mechanics and function. Excessive collagen deposition increases ventricular stiffness, impairs diastolic filling, disrupts electrical conduction pathways, and contributes to arrhythmogenesis [3]. Fibrotic remodeling also alters force transmission within cardiac tissue, potentially enhancing mechanosensitive signaling and further activating PIEZO-dependent pathways. This mechanobiological feedback loop is particularly important in chronic pressure overload conditions such as hypertension and aortic stenosis, where sustained mechanical stress progressively drives myocardial fibrosis and heart failure.
Recent studies have shown that PIEZO1 also contributes to matrix stiffness sensing in human atrial fibroblasts. Emig et al. [38] demonstrated that PIEZO1 regulates fibroblast mechanical properties and cellular adaptation to altered extracellular matrix conditions. Their findings suggest that PIEZO1 enables fibroblasts to directly detect changes in tissue rigidity and adjust their profibrotic behavior accordingly. Such stiffness-sensitive mechanotransduction mechanisms are likely central to the progression of fibrotic cardiovascular disease.
In addition to classical signaling pathways, emerging evidence suggests that PIEZO-mediated mechanotransduction interacts with epigenetic and epitranscriptomic regulatory networks controlling fibrotic gene expression. Garoffolo and Pesce [81] highlighted recent findings indicating that reductions in N6-methyladenosine (m6A) modification of Piezo2 RNA can influence cardiac fibrosis, suggesting an important connection between cellular mechanics and epitranscriptomic regulation. These studies indicate that mechanosensitive signaling pathways may regulate fibrosis not only through acute calcium-dependent signaling but also through long-term modulation of RNA stability, translation, and gene expression programs.
The interaction between mechanotransduction and epigenetic regulation represents an emerging frontier in cardiovascular mechanobiology. Mechanical forces can influence chromatin organization, transcription factor activation, RNA modification, and noncoding RNA signaling pathways involved in fibrosis and remodeling [3]. PIEZO channels may therefore function as upstream mechanosensitive regulators capable of integrating biomechanical stimuli with epigenetic control of fibroblast phenotype and extracellular matrix production.
PIEZO-mediated fibrotic remodeling is not limited to the myocardium. Similar mechanotransduction-dependent fibrotic processes have been described in vascular tissues, pulmonary fibroblasts, and other mechanically active organs [88]. These observations suggest that PIEZO channels may represent broadly conserved regulators of tissue fibrosis across multiple organ systems.
At the molecular level, PIEZO1 activity during fibrosis is influenced by membrane tension, extracellular matrix rigidity, cytoskeletal organization, and lipid membrane composition [21,62]. Progressive matrix stiffening alters force transmission to the plasma membrane and may lower the threshold for mechanosensitive activation. Consequently, fibrotic tissues may become increasingly sensitive to mechanical stimulation, thereby amplifying PIEZO-mediated signaling and accelerating disease progression.
The recognition of PIEZO1 as a central mediator of fibrosis and extracellular matrix remodeling has important therapeutic implications. Because fibrosis contributes significantly to heart failure progression, arrhythmias, and vascular dysfunction, targeting PIEZO-mediated mechanotransduction may represent a promising strategy for limiting pathological remodeling. Pharmacological modulation of PIEZO signaling could potentially reduce fibroblast activation, inflammatory signaling, and excessive matrix deposition while preserving normal adaptive tissue repair mechanisms [72].
Overall, current evidence strongly supports a critical role for PIEZO1-mediated mechanotransduction in fibrosis and extracellular matrix remodeling. Through regulation of inflammatory signaling, fibroblast activation, matrix stiffness sensing, and epigenetic pathways, PIEZO channels contribute to the progression of cardiovascular fibrosis and pathological remodeling. The dynamic interplay between mechanical stress, extracellular matrix stiffness, and mechanosensitive signaling forms a self-reinforcing cycle that drives disease progression, highlighting PIEZO1 as both a key mechanobiological regulator and a promising therapeutic target in fibrotic cardiovascular disease.
4.4. Ischemic Injury and Adverse Remodeling
Increasing evidence suggests that mechanotransduction pathways play important roles in myocardial ischemia processes by linking altered tissue mechanics to inflammatory signaling, calcium dysregulation, and cellular injury. Among the mechanosensitive pathways implicated in ischemic remodeling, PIEZO1 has emerged as a significant mediator of adverse cardiac responses to mechanical stress [31,40].
During myocardial ischemia and reperfusion, the heart experiences substantial alterations in mechanical loading conditions, including increased wall stress, tissue deformation, edema, extracellular matrix disruption, and changes in ventricular compliance [3]. These biomechanical disturbances alter membrane tension and activate mechanosensitive ion channels such as PIEZO1. Because PIEZO1 functions as a mechanically activated calcium-permeable cation channel, excessive activation during ischemic stress can profoundly influence intracellular calcium homeostasis and downstream injury pathways.
Recent work by Umbarkar et al. [40] provided direct evidence linking PIEZO1 to ischemia-induced adverse cardiac remodeling and dysfunction. Their study demonstrated that mechanosensitive PIEZO1 activation aggravates post-ischemic cardiac injury, ventricular remodeling, and functional decline. Importantly, suppression or modulation of PIEZO1 signaling attenuated adverse remodeling, suggesting that excessive mechanotransduction contributes directly to ischemic pathophysiology rather than merely reflecting secondary injury responses.
One of the major mechanisms through which PIEZO1 contributes to ischemic injury is calcium overload. Under physiological conditions, calcium signaling is tightly regulated to coordinate excitation-contraction coupling and cellular homeostasis. However, ischemia-reperfusion dramatically disrupts ionic balance, leading to excessive intracellular calcium accumulation [78]. Mechanical activation of PIEZO1 during ischemia may exacerbate this process by providing an additional pathway for calcium influx. Sustained calcium overload activates multiple pathological signaling cascades associated with contractile dysfunction, mitochondrial injury, and cardiomyocyte death.
Excessive intracellular calcium is strongly linked to oxidative stress generation during ischemic injury. Elevated calcium concentrations stimulate mitochondrial calcium uptake, disrupting oxidative phosphorylation and enhancing the production of reactive oxygen species (ROS). ROS accumulation damages proteins, lipids, and DNA while further impairing mitochondrial function and cellular viability. PIEZO1-mediated calcium dysregulation may therefore amplify oxidative stress and contribute to progressive myocardial injury following ischemia-reperfusion [39].
Mitochondrial dysfunction represents another critical consequence of abnormal PIEZO-mediated mechanotransduction during ischemic injury. Mitochondria are highly sensitive to calcium overload and oxidative stress, both of which can trigger opening of the mitochondrial permeability transition pore, loss of membrane potential, ATP depletion, and activation of apoptotic pathways. Because cardiomyocytes are heavily dependent on mitochondrial energy production, mitochondrial dysfunction significantly compromises contractile performance and accelerates heart failure progression. Emerging evidence suggests that PIEZO1-mediated mechanosensitive calcium influx contributes to mitochondrial injury under pathological mechanical stress conditions [40].
PIEZO1 activation may also promote multiple forms of cardiomyocyte cell death, including apoptosis, necrosis, and potentially ferroptosis or pyroptosis under severe ischemic conditions. Calcium overload activates calcium-dependent proteases such as calpains, which degrade cytoskeletal and contractile proteins and contribute to irreversible cellular injury [39]. Simultaneously, oxidative stress and mitochondrial dysfunction activate apoptotic signaling pathways, further exacerbating myocardial damage. These mechanisms collectively contribute to infarct expansion, ventricular thinning, and progressive deterioration of cardiac function.
Inflammatory activation is another major component of ischemia-induced adverse remodeling influenced by PIEZO-mediated mechanotransduction. Following myocardial infarction, damaged cardiomyocytes release inflammatory mediators and danger-associated molecular patterns that recruit immune cells and activate fibroblasts [36]. Mechanical stress associated with infarct expansion and altered ventricular loading further amplifies inflammatory signaling through mechanosensitive pathways. PIEZO1 activation has been linked to production of inflammatory cytokines such as interleukin-6 and activation of stress-responsive pathways, including p38 MAPK signaling [37]. Persistent inflammatory activation contributes to extracellular matrix remodeling, fibrosis, ventricular stiffening, and progression toward chronic heart failure.
The interaction between ischemic injury and fibrosis is particularly important in post-infarction remodeling. Following myocardial infarction, fibroblasts become activated and deposit extracellular matrix proteins to stabilize damaged tissue and form scar tissue. However, excessive fibrotic remodeling impairs ventricular compliance and electrical conduction, contributing to diastolic dysfunction and arrhythmogenesis [3]. Because PIEZO1 regulates both cardiomyocyte stress responses and fibroblast mechanotransduction, abnormal PIEZO signaling may coordinate multiple aspects of adverse remodeling following ischemic injury.
Mechanical alterations in the infarcted myocardium further enhance mechanosensitive signaling. Ischemic injury changes tissue stiffness, extracellular matrix composition, and wall stress distribution, creating abnormal biomechanical environments that can chronically activate PIEZO-dependent pathways [2]. This creates a self-perpetuating cycle in which mechanical dysfunction promotes mechanotransduction signaling, further exacerbating inflammation, fibrosis, and ventricular remodeling.
Emerging studies also suggest that PIEZO-mediated ischemic injury may interact with neurohumoral and autonomic signaling pathways involved in heart failure progression. Mechanical stress influences sympathetic activation, renin-angiotensin signaling, and inflammatory responses, all of which contribute to adverse remodeling after myocardial infarction [89]. PIEZO1 may therefore function as an important integrator linking altered cardiac mechanics to systemic cardiovascular stress responses.
At the molecular level, PIEZO1 activity during ischemia-reperfusion is likely influenced by membrane lipid composition, oxidative stress, cytoskeletal disruption, and extracellular matrix remodeling [21,62]. Ischemic injury alters membrane fluidity and cellular mechanics, potentially modifying PIEZO activation thresholds and mechanosensitivity. Such changes may enhance susceptibility to pathological calcium influx during reperfusion and mechanical overload.
The growing recognition of PIEZO1 as a contributor to ischemia-induced remodeling has important therapeutic implications. Pharmacological modulation of PIEZO-mediated mechanotransduction may represent a novel strategy for reducing calcium overload, oxidative stress, inflammation, and adverse remodeling following myocardial infarction. Targeting mechanosensitive signaling pathways could potentially complement existing reperfusion and neurohumoral therapies to improve post-infarction recovery and limit heart failure progression [72].
Overall, accumulating evidence indicates that PIEZO1-mediated mechanotransduction plays an important role in ischemic injury and adverse cardiac remodeling. Mechanical stress during ischemia and reperfusion activates PIEZO-dependent calcium signaling pathways that contribute to oxidative stress, mitochondrial dysfunction, inflammatory activation, cell death, and fibrosis. By linking altered tissue mechanics to pathological intracellular signaling, PIEZO1 functions as a critical mediator of post-ischemic remodeling and represents a promising therapeutic target for mitigating ischemia-induced cardiovascular injury and heart failure progression.
4.5. Vascular Inflammation and Atherosclerosis
Among the mechanosensitive pathways involved in vascular inflammation, PIEZO1 has emerged as an important mediator linking abnormal hemodynamic stress to endothelial dysfunction, immune activation, and atherogenesis [28,73].
Under physiological conditions, endothelial cells experience laminar shear stress generated by steady blood flow. Laminar flow promotes endothelial quiescence, nitric oxide production, anti-inflammatory signaling, and vascular homeostasis [1]. In contrast, disturbed flow patterns commonly occur at arterial bifurcations, curvatures, and regions downstream of stenotic lesions, where blood flow becomes oscillatory or turbulent. These abnormal hemodynamic conditions generate irregular mechanical forces that activate inflammatory signaling pathways within endothelial cells and circulating immune cells, thereby initiating atherogenic remodeling [2].
PIEZO1 functions as a primary endothelial mechanosensor capable of detecting alterations in fluid shear stress and membrane tension [29]. Under disturbed flow conditions, abnormal PIEZO1 activation alters calcium-dependent signaling pathways that regulate endothelial function, inflammatory responses, and vascular remodeling [73]. Mechanical activation of PIEZO1 influences endothelial nitric oxide signaling, cytoskeletal organization, oxidative stress responses, and expression of inflammatory mediators, thereby contributing to vascular dysfunction and atherogenesis.
One important mechanism linking PIEZO1 to vascular inflammation involves activating inflammatory signaling pathways in endothelial cells. Disturbed shear stress can induce endothelial expression of adhesion molecules, cytokines, and chemokines that recruit circulating leukocytes to the vascular wall [1]. PIEZO1-mediated calcium influx may amplify these responses by activating stress-sensitive signaling pathways, such as MAPK and NF-κB, and by promoting inflammatory cytokine production [27]. Chronic endothelial inflammation promotes increased vascular permeability, leukocyte adhesion, oxidative stress, and extracellular matrix remodeling, all of which contribute to plaque initiation and progression.
Beyond endothelial cells, PIEZO1 also regulates inflammatory activation in circulating monocytes and immune cells. Monocytes are highly responsive to biomechanical forces encountered during circulation and vascular injury. Mechanical stimulation through PIEZO1 can activate inflammatory pathways within monocytes, promoting cytokine production and inflammatory phenotypes associated with vascular disease. Baratchi et al. [42] demonstrated that elevated shear stress associated with aortic stenosis promotes PIEZO1-mediated monocyte activation and inflammation. Importantly, the reduction of abnormal shear stress following transcatheter aortic valve implantation (TAVI) significantly attenuated monocyte activation and inflammatory signaling. These findings provided direct evidence that pathological hemodynamic forces regulate immune cell activation through PIEZO1-dependent mechanotransduction pathways.
The study by Baratchi et al. [42] highlighted the important relationship between altered cardiovascular biomechanics and systemic inflammation. Severe aortic stenosis generates extremely high shear stress within the circulation, exposing both endothelial cells and circulating blood cells to abnormal mechanical loading. PIEZO1-mediated sensing of these pathological forces contributes to inflammatory activation and vascular dysfunction. Restoration of more physiological flow patterns following valve replacement reduced mechanosensitive inflammatory signaling, suggesting that normalization of hemodynamic forces can directly improve inflammatory cardiovascular responses.
Disturbed flow-induced PIEZO1 activation may additionally contribute to oxidative stress, another major driver of atherosclerosis. Abnormal mechanotransduction increases production of reactive oxygen species (ROS), which impair nitric oxide bioavailability, damage endothelial structures, and promote inflammatory signaling [26]. Oxidative stress also oxidizes low-density lipoproteins (LDL), enhancing macrophage foam cell formation and accelerating plaque development. Through regulation of calcium signaling and inflammatory pathways, PIEZO1 may therefore function as an upstream mediator of oxidative vascular injury.
Mechanical stress and vascular inflammation further contribute to extracellular matrix remodeling and vascular stiffening. Chronic inflammatory activation stimulates smooth muscle cell proliferation, collagen deposition, and matrix degradation within the arterial wall [2]. Progressive vascular stiffening alters local hemodynamic forces and amplifies mechanosensitive signaling, creating a self-reinforcing cycle of abnormal flow, inflammation, and remodeling. Because PIEZO1 responds directly to membrane tension and shear stress, vascular stiffening may further enhance pathological mechanotransduction and accelerate atherosclerotic progression.
Emerging evidence also suggests that PIEZO-mediated vascular inflammation may contribute to peripheral arterial disease (PAD). Zhao et al. [44] reported that PIEZO1 expression is upregulated in peripheral arterial disease and in murine models of vascular ischemia. Increased mechanosensitive signaling under conditions of disturbed flow and vascular injury may therefore contribute broadly to inflammatory vascular diseases beyond coronary atherosclerosis.
At the molecular level, PIEZO1-mediated inflammatory signaling is influenced by membrane mechanics, cytoskeletal organization, extracellular matrix stiffness, and local flow conditions [21,62]. Disturbed flow patterns alter endothelial membrane tension and cytoskeletal dynamics, potentially modifying PIEZO activation thresholds and inflammatory responses. Chronic inflammatory conditions additionally change vascular stiffness and extracellular matrix composition, further influencing mechanotransduction signaling pathways.
Recent studies have also begun to explore the interaction between PIEZO signaling and immune mechanobiology. Mechanical forces regulate not only endothelial cells but also macrophages, monocytes, lymphocytes, and other immune cells involved in vascular inflammation [90]. PIEZO channels may therefore coordinate mechanosensitive inflammatory responses across multiple cell populations within the vascular microenvironment.
The growing recognition of PIEZO1 as a regulator of vascular inflammation and atherogenesis has important therapeutic implications. Because abnormal mechanotransduction contributes directly to endothelial dysfunction, immune activation, and vascular remodeling, targeting PIEZO-mediated signaling may represent a novel strategy for limiting atherosclerosis and inflammatory cardiovascular disease. Pharmacological modulation of PIEZO1 could potentially reduce inflammatory activation, improve endothelial function, and normalize vascular responses to disturbed flow [72]. However, given the essential physiological roles of PIEZO1 in vascular homeostasis, selective and context-dependent therapeutic approaches will likely be necessary.
Overall, current evidence strongly supports a critical role for PIEZO1-mediated mechanotransduction in vascular inflammation and atherosclerosis. Disturbed flow patterns activate mechanosensitive signaling pathways that promote endothelial dysfunction, monocyte activation, oxidative stress, inflammatory signaling, and vascular remodeling. By linking abnormal hemodynamic forces to cellular inflammatory responses, PIEZO1 functions as a central mechanobiological regulator of atherogenesis and vascular disease progression. Continued investigation of PIEZO-dependent vascular inflammation may provide important insights into cardiovascular disease mechanisms and identify new therapeutic opportunities for preventing atherosclerosis and inflammatory vascular disorders.
4.6. Translational and Human Cardiovascular Evidence
Although much of the current mechanistic understanding of PIEZO signaling derives from experimental cellular and rodent studies, increasing human evidence supports the translational relevance of mechanosensitive ion channels in cardiovascular disease. Darkow et al. performed a meta-analysis of mechanosensitive ion channel expression in human hearts and identified chamber-specific and disease-associated expression patterns involving PIEZO-related mechanotransduction pathways [91]. Their findings suggest important roles for mechanosensitive signaling in atrial remodeling, ventricular dysfunction, arrhythmogenesis, and the progression of heart failure.
Human atrial fibroblast studies have additionally demonstrated that PIEZO1 contributes to matrix stiffness sensing and inflammatory remodeling under pathological mechanical environments. Clinical observations further suggest altered mechanosensitive signaling in hypertension, vascular inflammation, ischemic injury, and fibrotic remodeling. These findings collectively support the clinical relevance of PIEZO-mediated cardiovascular mechanotransduction while highlighting the need for additional patient-specific mechanobiological investigations and translational cardiovascular studies.
4.7. Systemic Consequences of PIEZO Dysfunction
Hereditary xerocytosis represents an important systemic consequence of PIEZO1 dysfunction rather than a primary cardiovascular disease. Gain-of-function mutations in PIEZO1 alter erythrocyte mechanosensitive ion transport, resulting in abnormal calcium influx, erythrocyte dehydration, impaired membrane deformability, and chronic hemolytic anemia. Picard et al. analyzed a large retrospective cohort of patients with PIEZO1-hereditary xerocytosis and demonstrated substantial clinical and biological heterogeneity associated with dysregulated mechanosensitive ion transport [92]. Although primarily hematologic in presentation, hereditary xerocytosis highlights the broad physiological importance of PIEZO-mediated mechanotransduction and the systemic consequences of abnormal mechanosensitive signaling.
Table 2 summarizes representative cardiovascular diseases associated with abnormal PIEZO signaling, including implicated cell types, downstream molecular pathways, and major pathological consequences.
Table 2. PIEZO-mediated cardiovascular diseases, implicated cell types, signaling pathways, and pathological consequences.
|
Disease/Condition |
Major Cell Type |
PIEZO Isoform |
Key Downstream Pathways |
Representative Mechanisms |
Pathological Consequences |
|---|---|---|---|---|---|
|
Hypertension |
Endothelial cells |
PIEZO1 |
eNOS/NO signaling, NF-κB, calcium signaling |
Altered shear stress sensing, endothelial dysfunction, impaired nitric oxide bioavailability |
Increased vascular stiffness, elevated vascular resistance |
|
Atherosclerosis and vascular inflammation |
Endothelial cells, monocytes/macrophages |
PIEZO1 |
NF-κB, inflammatory cytokines, oxidative stress pathways |
Disturbed flow-mediated inflammatory activation and leukocyte recruitment |
Plaque progression and vascular remodeling |
|
Cardiac hypertrophy |
Cardiomyocytes |
PIEZO1 |
Calcineurin/NFAT, MAPK, CaMKII |
Pressure overload-induced calcium influx and hypertrophic gene activation |
Pathological myocardial remodeling |
|
Heart failure |
Cardiomyocytes |
PIEZO1 |
Calcium dysregulation, stress signaling pathways |
Chronic mechanosensitive calcium overload and maladaptive remodeling |
Contractile dysfunction and heart failure progression |
|
Cardiac fibrosis |
Cardiac fibroblasts |
PIEZO1 |
IL-6, p38 MAPK, TGF-β/Smad |
Fibroblast activation, matrix stiffness sensing, extracellular matrix deposition |
Myocardial stiffening and impaired diastolic function |
|
Ischemic injury |
Cardiomyocytes |
PIEZO1 |
Oxidative stress, mitochondrial dysfunction, calcium overload |
Ischemia-induced mechanosensitive injury and inflammatory remodeling |
Cardiomyocyte death and adverse remodeling |
|
HFpEF |
Cardiomyocytes, fibroblasts |
PIEZO1 |
Calcium signaling, profibrotic pathways |
Altered mechanotransduction and ventricular stiffening |
Diastolic dysfunction |
|
Peripheral arterial disease |
Endothelial cells, vascular smooth muscle cells |
PIEZO1 |
Shear stress signaling, inflammatory pathways |
Abnormal vascular mechanosensing and impaired flow adaptation |
Vascular dysfunction and ischemia |
|
Lymphatic remodeling |
Lymphatic endothelial cells |
PIEZO1 |
Flow-sensitive calcium signaling |
Impaired lymphatic mechanotransduction and vessel remodeling |
Abnormal lymphatic expansion |
|
Hereditary xerocytosis * |
Erythrocytes |
PIEZO1 |
Mechanosensitive ion transport, calcium influx |
Gain-of-function mutations altering erythrocyte deformability |
Hemolytic anemia and systemic mechanotransduction abnormalities |
* Hereditary xerocytosis is included as a representative systemic consequence of PIEZO dysfunction rather than a primary cardiovascular disease.
Collectively, these findings demonstrate that PIEZO-mediated mechanotransduction influences multiple aspects of cardiovascular physiology and pathology through integrated calcium-dependent signaling pathways and biomechanical sensing mechanisms. The broad involvement of PIEZO channels in vascular remodeling, inflammation, fibrosis, and myocardial adaptation has therefore stimulated growing interest in therapeutic modulation of mechanosensitive signaling pathways.
5. Pharmacological Targeting of PIEZO Channels
Increasing evidence suggests that PIEZO-mediated mechanotransduction plays important roles in hypertension, cardiac hypertrophy, fibrosis, ischemic injury, vascular inflammation, and heart failure progression, making PIEZO channels attractive therapeutic targets [26,28]. However, selective pharmacological targeting remains challenging because PIEZO channels are widely expressed in endothelial cells, cardiomyocytes, fibroblasts, erythrocytes, neurons, immune cells, and smooth muscle cells, where they regulate essential physiological functions [27,51,56,93].
Current therapeutic approaches include direct channel inhibitors, mechanosensitivity modulators, membrane tension modifiers, and downstream signaling inhibitors. GsMTx4, a mechanosensitive ion channel-modulating peptide, suppresses mechanically activated calcium influx and may reduce pathological calcium overload and mechanically induced cellular injury [72]. Yoda1, a synthetic PIEZO1 agonist, and Dooku1, an antagonist of Yoda1-mediated activation, have also become important pharmacological tools for investigating PIEZO regulation [13,60].
Because PIEZO channels are directly gated by membrane tension, alterations in membrane lipid composition, cholesterol content, cytoskeletal organization, and extracellular matrix stiffness can significantly influence channel activation [14,20,62]. Consequently, modulation of membrane mechanics may indirectly regulate pathological mechanotransduction. In addition, targeting downstream signaling pathways activated by excessive PIEZO-mediated calcium influx, including calcineurin/NFAT, MAPK, IL-6, oxidative stress, and inflammatory signaling pathways, may provide therapeutic benefit without directly inhibiting PIEZO channels themselves [37,39,40,78].
Future therapeutic development will likely require an improved understanding of tissue-specific PIEZO regulation and mechanotransduction dynamics. Continued advances in structural biology, cryo-electron microscopy, molecular simulations, and computational modeling may facilitate the development of more selective mechanotransduction-based therapies for cardiovascular disease.
Several important clinical applications of PIEZO-targeted therapies are currently being explored or proposed:
-
-
Anti-hypertrophic Therapies
Because PIEZO1 contributes to pressure overload-induced hypertrophic signaling and calcium dysregulation, selective inhibition may help prevent maladaptive cardiac remodeling and progression to heart failure [35,39]. Modulating mechanosensitive calcium influx could potentially reduce activation of calpain, calcineurin/NFAT, and CaMKII pathways involved in pathological hypertrophy.
-
-
Anti-fibrotic Interventions
PIEZO1-mediated mechanotransduction in fibroblasts promotes extracellular matrix deposition, inflammatory activation, and tissue stiffening [37,38]. Targeting these pathways may reduce myocardial fibrosis, improve ventricular compliance, and limit adverse remodeling following hypertension or myocardial infarction.
-
-
Vascular Protective Agents
Endothelial PIEZO1 regulates vascular tone, nitric oxide signaling, and adaptation to shear stress [29,30]. Pharmacological modulation of endothelial mechanotransduction may therefore improve endothelial function, reduce vascular inflammation, and attenuate progression of hypertension and atherosclerosis.
-
-
Anti-inflammatory Treatments
PIEZO-mediated mechanotransduction contributes to inflammatory signaling in endothelial cells, fibroblasts, and circulating immune cells [42]. Inhibition of pathological mechanosensitive inflammatory activation may reduce vascular inflammation, atherogenesis, and post-ischemic remodeling.
-
-
Ischemia-Reperfusion Protection
Because excessive PIEZO activation contributes to calcium overload, oxidative stress, mitochondrial dysfunction, and cardiomyocyte death during ischemia-reperfusion injury, mechanosensitive channel modulation may provide cardioprotective effects following myocardial infarction [40].
Emerging technologies such as artificial intelligence, machine learning, and high-throughput computational screening may further accelerate PIEZO-targeted drug discovery. Deep learning-assisted molecular modeling and kinetic simulations may help identify novel modulators capable of selectively altering channel gating behavior or mechanosensitivity under pathological conditions. Computational approaches integrating structural biology, electrophysiology, and pharmacodynamics may prove especially valuable given the complexity of PIEZO channel mechanics and multimodal regulation.
Despite substantial progress, several major challenges remain. PIEZO channels exhibit highly complex gating behavior influenced by membrane tension, lipid composition, cytoskeletal interactions, extracellular matrix mechanics, and voltage dependence [22,23]. Achieving tissue-specific modulation without disrupting essential physiological functions will require increasingly sophisticated pharmacological strategies. Furthermore, long-term effects of PIEZO modulation on cardiovascular adaptation and systemic mechanobiology remain incompletely understood.
Overall, PIEZO channels represent highly promising but technically challenging therapeutic targets for cardiovascular disease. Advances in structural biology, mechanobiology, pharmacology, and computational modeling are rapidly improving understanding of PIEZO channel regulation and therapeutic potential. Continued interdisciplinary research will be essential for translating mechanotransduction discoveries into clinically effective therapies that target pathological mechanical signaling while preserving normal cardiovascular function.
6. Emerging Technologies and Future Directions
6.1. Machine Learning and Artificial Intelligence
Machine learning and artificial intelligence (AI) are increasingly transforming biomedical research by enabling the analysis of large, complex, and multidimensional datasets that are often difficult to interpret using conventional analytical methods. In mechanobiology and ion channel research, these computational approaches have become particularly valuable because mechanosensitive signaling involves highly dynamic, nonlinear, and stochastic processes occurring across multiple spatial and temporal scales [51,59]. The study of PIEZO channels presents especially significant analytical challenges due to the complexity of mechanogating behavior, rapid activation and inactivation kinetics, hidden conformational states, and substantial experimental noise associated with electrophysiological recordings. Consequently, machine learning approaches are emerging as powerful tools for advancing understanding of PIEZO-mediated mechanotransduction and cardiovascular mechanobiology.
Traditional electrophysiological analysis methods often rely on manual event detection, curve fitting, or hidden Markov modeling to interpret ion channel recordings. While these approaches have contributed significantly to understanding mechanosensitive channels, they become increasingly limited when applied to large patch-clamp datasets containing stochastic gating events, variable transition kinetics, and noisy single-channel signals [55]. PIEZO channels exhibit particularly complex kinetic behavior because their activation depends not only on electrical conditions but also on membrane tension, lipid mechanics, cytoskeletal interactions, extracellular matrix stiffness, and force-dependent conformational transitions [14,19]. These complexities make computational analysis both challenging and computationally intensive.
Machine learning algorithms provide important advantages in this context because they can identify hidden patterns, nonlinear relationships, and time-dependent transitions within large electrophysiological datasets. Deep learning approaches such as convolutional neural networks (CNNs), recurrent neural networks (RNNs), recurrent convolutional neural networks (RCNNs), and long short-term memory (LSTM) architectures are especially promising for studying PIEZO gating kinetics and mechanotransduction dynamics. These models are well suited for analyzing time-series data because they can learn temporal dependencies and identify recurring activation patterns associated with hidden ion channel states.
Patch-clamp electrophysiology generates large volumes of high-frequency current recordings containing transient mechanosensitive channel events. Deep learning algorithms can process these datasets more rapidly and accurately than traditional manual approaches while reducing user bias and improving reproducibility. RCNN and LSTM architectures are particularly advantageous because they combine feature extraction capabilities with temporal sequence analysis, enabling identification of complex gating transitions and hidden kinetic states from noisy recordings.
Recent studies in ion channel analysis have demonstrated the effectiveness of deep learning frameworks for event detection and kinetic modeling. For example, neural-network-based approaches such as Deep-Channel have shown that machine learning can identify single-molecule ion channel events with high accuracy and speed compared with traditional hidden Markov methods. Similar computational approaches may prove especially valuable for PIEZO channels because mechanosensitive gating involves force-dependent conformational transitions that are often difficult to resolve experimentally.
Beyond electrophysiological analysis, artificial intelligence is increasingly being integrated with structural biology and mechanobiological simulations. Advances in cryo-electron microscopy have generated detailed structural models of PIEZO channels that reveal their distinctive curved membrane architecture and force-sensing mechanisms [16,17]. Machine learning algorithms can now be combined with molecular dynamics simulations and biophysical modeling to predict conformational changes, gating transitions, ion permeation properties, and force-dependent structural responses [69,70]. These computational approaches provide new opportunities to investigate how membrane tension, lipid composition, cytoskeletal organization, and extracellular matrix mechanics influence PIEZO activation.
One of the most promising future directions involves AI-assisted ion channel state prediction. Mechanosensitive ion channels often transition between multiple hidden conformational states that cannot be directly observed experimentally. Deep learning algorithms may help reconstruct these hidden states from electrophysiological recordings and predict force-dependent gating transitions with greater accuracy than traditional statistical approaches. Such predictive models could significantly improve understanding of PIEZO kinetics under physiological and pathological mechanical conditions.
Another important emerging area is multiscale mechanobiological modeling. Mechanotransduction involves interactions across molecular, cellular, tissue, and organ scales, making integrative computational approaches essential for understanding cardiovascular biomechanics. AI-driven multiscale models may combine electrophysiological data, structural simulations, fluid dynamics, membrane mechanics, and cellular signaling networks into unified mechanobiological frameworks. These approaches could help explain how microscopic PIEZO channel activation influences large-scale cardiovascular remodeling, vascular adaptation, and disease progression.
Machine learning may also enable personalized mechanotransduction analysis and precision cardiovascular medicine. Individual differences in genetics, extracellular matrix composition, vascular stiffness, and cellular mechanics likely influence mechanosensitive signaling and susceptibility to cardiovascular disease. AI-based computational models integrating patient-specific imaging, electrophysiology, genomic data, and biomechanical parameters may eventually allow personalized prediction of mechanotransduction responses and disease risk. Such approaches could identify patients at increased risk for hypertension, fibrosis, cardiac hypertrophy, or mechanotransduction-related cardiovascular dysfunction.
Drug discovery and pharmacological screening represent another highly promising application of artificial intelligence in PIEZO research. Because PIEZO channels exhibit highly complex gating behavior influenced by membrane tension and biomechanical context, conventional drug discovery approaches remain challenging [72]. AI-assisted screening platforms may accelerate identification of compounds capable of modulating PIEZO activity by predicting ligand binding, gating effects, and mechanosensitivity alterations. Deep learning models can analyze large chemical libraries, simulate channel-drug interactions, and identify candidate compounds with improved selectivity and efficacy.
Machine learning may also facilitate the development of computational “digital twins” of cardiovascular mechanotransduction systems. These virtual physiological models could integrate biomechanical forces, electrophysiology, molecular signaling, and tissue remodeling into patient-specific simulations that predict disease progression and therapeutic responses. Such technologies may eventually support clinical decision-making and individualized treatment strategies for mechanotransduction-related cardiovascular diseases.
Despite these exciting advances, several important challenges remain. AI models require large, high-quality datasets for training and validation, yet mechanobiological data are often noisy, heterogeneous, and experimentally difficult to standardize. In addition, deep learning algorithms can function as “black boxes”, making biological interpretation of learned features difficult. Future progress will therefore require improved integration between experimental mechanobiology, computational modeling, and explainable AI frameworks.
Another major challenge involves the inherently multiscale nature of mechanotransduction. PIEZO channel behavior depends on interactions among membrane lipids, cytoskeletal elements, extracellular matrix mechanics, tissue stiffness, and hemodynamic forces [62]. Accurately incorporating these factors into computational models remains computationally demanding and biologically complex. Continued advances in high-performance computing, physics-informed neural networks, and multimodal data integration will likely be essential for overcoming these limitations.
Overall, machine learning and artificial intelligence are rapidly reshaping the study of PIEZO channels and cardiovascular mechanobiology. By enabling analysis of complex electrophysiological datasets, prediction of hidden ion channel states, multiscale mechanobiological modeling, personalized mechanotransduction analysis, and AI-assisted drug discovery, these computational technologies offer unprecedented opportunities to advance understanding of mechanosensitive signaling. Continued integration of artificial intelligence with structural biology, electrophysiology, and cardiovascular research is expected to play a transformative role in future mechanobiology and therapeutic development.
6.2. Multiscale Cardiovascular Mechanobiology
Cardiovascular mechanobiology is inherently a multiscale phenomenon involving complex interactions among molecular structures, cellular signaling pathways, tissue biomechanics, organ-level hemodynamics, and systemic physiological regulation. Mechanical forces generated by blood flow, pressure, myocardial contraction, and extracellular matrix deformation are sensed at the molecular level yet ultimately influence whole-organ cardiovascular function and disease progression [1,2]. Because PIEZO-mediated mechanotransduction operates across these interconnected biological scales, future advances in cardiovascular research will require integrated multiscale approaches that link molecular force sensing to tissue remodeling and systemic pathology.
At the molecular level, mechanotransduction begins with force-dependent conformational changes in mechanosensitive proteins such as PIEZO channels. Structural studies using cryo-electron microscopy have revealed the unique curved architecture of PIEZO channels and clarified how membrane tension induces pore opening and ion permeation [16,17]. Molecular dynamics simulations and biophysical modeling have further demonstrated how membrane curvature, lipid composition, cytoskeletal interactions, and extracellular forces regulate PIEZO gating behavior [69,70]. These nanoscale structural mechanisms form the foundation of mechanotransduction signaling throughout the cardiovascular system.
At the cellular level, PIEZO-mediated calcium influx activates complex intracellular signaling pathways involved in endothelial adaptation, cardiomyocyte hypertrophy, fibroblast activation, inflammatory signaling, and vascular remodeling [32,39]. Endothelial cells respond to shear stress through nitric oxide signaling and cytoskeletal reorganization, cardiomyocytes adapt to mechanical loading through hypertrophic signaling pathways, and fibroblasts regulate extracellular matrix remodeling in response to tissue stiffness [37,38]. These cellular responses are highly interconnected and dynamically influenced by local biomechanical conditions.
At the tissue level, mechanical forces regulate extracellular matrix organization, vascular stiffness, myocardial compliance, and electrical conduction properties. Chronic alterations in tissue biomechanics contribute to hypertension, fibrosis, atherosclerosis, ventricular remodeling, and heart failure progression [3]. Importantly, tissue-level changes such as matrix stiffening can further amplify mechanotransduction signaling by increasing force transmission to cellular membranes, thereby creating self-reinforcing cycles of pathological remodeling. PIEZO-mediated mechanotransduction therefore functions not only as a cellular sensing mechanism but also as a regulator of tissue-scale biomechanical adaptation.
At the organ level, cardiovascular mechanobiology influences ventricular function, vascular resistance, hemodynamic regulation, coronary perfusion, and systemic blood pressure homeostasis. Abnormal mechanical loading conditions associated with hypertension, valvular disease, myocardial infarction, and vascular stenosis alter hemodynamic forces throughout the cardiovascular system and activate widespread mechanotransduction responses [76]. For example, disturbed blood flow patterns promote endothelial inflammation and atherosclerosis, while pressure overload drives hypertrophic remodeling and ventricular dysfunction [35,42]. Understanding how molecular mechanosensors contribute to these organ-level pathologies requires integrative multiscale analysis.
Future research in cardiovascular mechanobiology will therefore require comprehensive frameworks capable of integrating molecular, cellular, tissue, and organ-level mechanotransduction processes into unified mechanistic models. Combining structural biology, computational modeling, biomechanics, and systems biology will be essential for understanding how mechanical forces influence cardiovascular physiology and disease progression across scales.
Figure 5 illustrates a multiscale framework of cardiovascular mechanotransduction linking nanoscale membrane mechanics to organ-level cardiovascular disease progression. Mechanical forces such as shear stress, cyclic stretch, hydrostatic pressure, and extracellular matrix stiffness initially act at the nanoscale level, where they induce membrane deformation, alter lipid tension, and activate PIEZO channels embedded within the plasma membrane. Mechanical activation of PIEZO channels promotes calcium-permeable cation influx and initiates intracellular mechanotransduction signaling pathways. At the cellular scale, these signaling mechanisms regulate endothelial adaptation, nitric oxide production, inflammatory activation, cardiomyocyte hypertrophy, fibroblast differentiation, and mechanosensitive calcium homeostasis. Persistent or dysregulated mechanotransduction subsequently contributes to tissue-level remodeling processes, including extracellular matrix deposition, fibrosis, vascular stiffening, altered tissue biomechanics, and inflammatory remodeling. These pathological changes progressively impair cardiovascular structure and function at the organ level, contributing to hypertension, cardiac hypertrophy, ischemic injury, vascular inflammation, atherosclerosis, and heart failure progression. Collectively, Figure 5 highlights the interconnected and hierarchical nature of mechanotransduction across molecular, cellular, tissue, and organ scales, emphasizing the importance of integrated multiscale approaches for understanding cardiovascular disease mechanisms and therapeutic development.
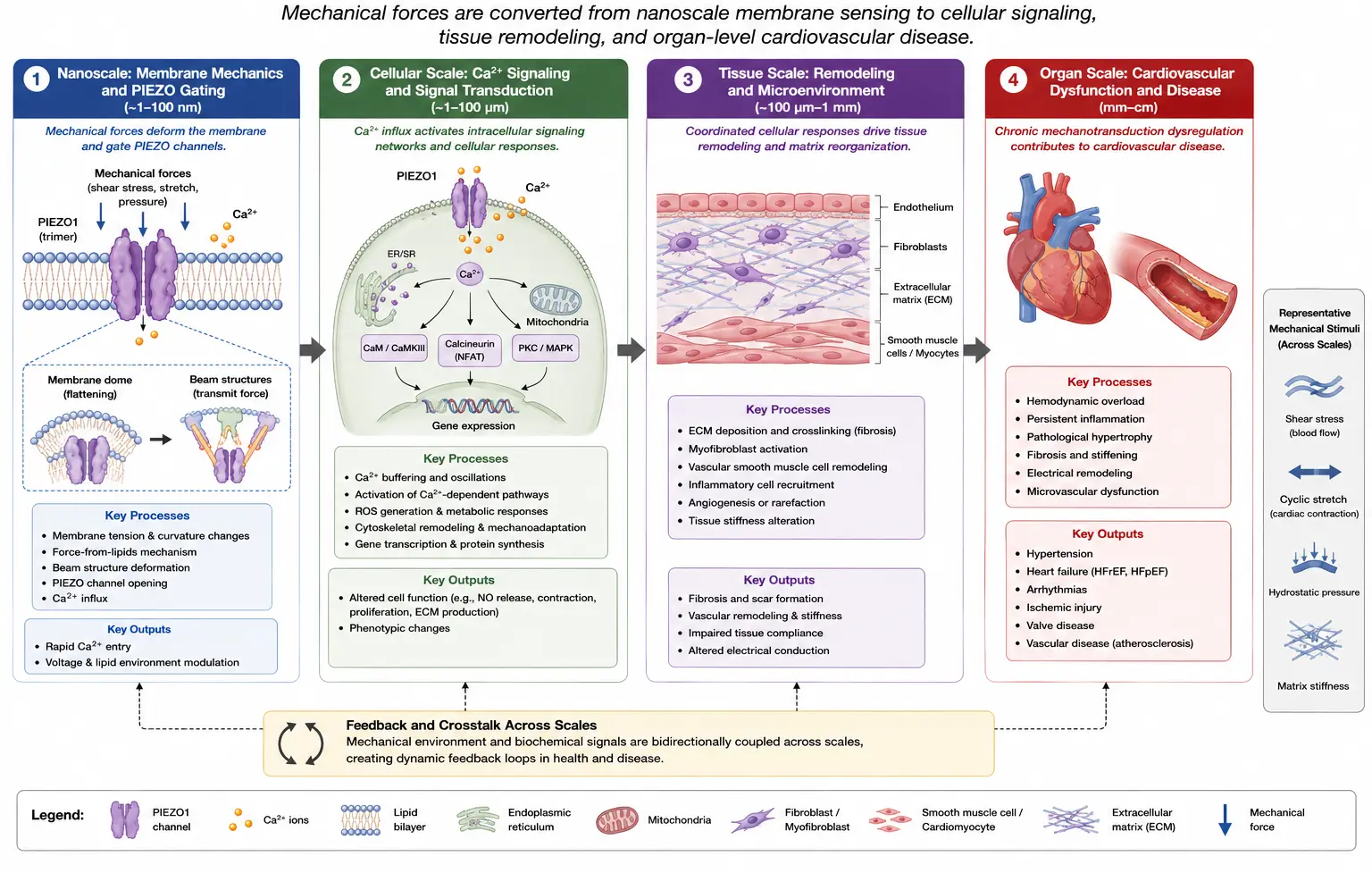
Figure 5. Multiscale cardiovascular mechanotransduction framework. Schematic illustration of the hierarchical organization of cardiovascular mechanotransduction across multiple biological scales. Mechanical forces acting at the nanoscale induce membrane deformation, lipid tension, and force-dependent activation of PIEZO channels embedded within the cellular membrane. PIEZO-mediated calcium influx subsequently initiates intracellular signaling pathways involved in endothelial adaptation, cardiomyocyte hypertrophy, inflammatory activation, and fibroblast mechanotransduction at the cellular scale. These signaling processes contribute to tissue-level extracellular matrix remodeling, fibrosis, vascular stiffening, and altered biomechanical properties. Progressive remodeling ultimately drives organ-level cardiovascular dysfunction, including hypertension, cardiac hypertrophy, ischemic injury, vascular inflammation, and heart failure. The figure highlights the interconnected relationships among membrane mechanics, cellular signaling, tissue remodeling, and systemic cardiovascular disease progression.
Structural biology will continue to play a foundational role in these efforts. Advances in cryo-electron microscopy, super-resolution imaging, and molecular biophysics are providing increasingly detailed insights into PIEZO channel structure, gating mechanisms, and force transmission pathways [18,19]. These structural data can be integrated with molecular simulations and computational biomechanics to predict how mechanical stimuli influence channel behavior under physiological and pathological conditions.
Computational modeling represents another critical component of multiscale mechanobiology. Finite element modeling, fluid-structure interaction simulations, molecular dynamics, and multiscale electrophysiological models can help bridge the gap between molecular mechanosensing and tissue-level cardiovascular function [69]. Computational fluid dynamics (CFD) may be particularly useful for investigating how altered blood flow patterns influence endothelial PIEZO activation, vascular inflammation, and atherogenesis. Similarly, biomechanical models of myocardial stress distribution can help clarify how pressure overload and ventricular remodeling influence cardiomyocyte mechanotransduction and the progression of heart failure.
Systems biology approaches will also be essential for understanding the highly interconnected signaling networks involved in cardiovascular mechanotransduction. Mechanical forces influence not only ion channels but also inflammatory signaling, metabolic regulation, epigenetic pathways, extracellular matrix remodeling, and neurohumoral activation [81]. Integrating transcriptomics, proteomics, metabolomics, and mechanobiological data may help identify global regulatory networks linking biomechanical stress to cardiovascular disease.
Artificial intelligence and machine learning are expected to become increasingly important for multiscale mechanobiological research. AI-driven computational frameworks can integrate large and heterogeneous datasets across multiple biological scales, including structural imaging, electrophysiology, biomechanics, genomics, and clinical data [59]. Such approaches may facilitate predictive modeling of mechanotransduction pathways and identification of patient-specific disease mechanisms.
An important future direction involves the development of “digital twin” cardiovascular models that incorporate personalized biomechanical and molecular data. These computational representations of individual patients could potentially simulate hemodynamics, mechanotransduction signaling, tissue remodeling, and therapeutic responses under various clinical conditions. Personalized mechanobiological models may ultimately support precision medicine approaches for hypertension, heart failure, fibrosis, and vascular disease.
Multiscale mechanobiology will also be important for advancing translational and therapeutic research. Pharmacological targeting of PIEZO-mediated mechanotransduction requires understanding how molecular interventions influence cellular signaling, tissue remodeling, and organ-level cardiovascular function [72]. Without multiscale integration, therapeutic modulation of mechanosensitive pathways may produce unintended systemic consequences due to the widespread physiological roles of mechanotransduction.
Experimental advances in tissue engineering and organ-on-chip technologies are additionally expected to enhance multiscale cardiovascular research. Biomimetic systems incorporating controlled mechanical loading, engineered extracellular matrices, and microfluidic flow conditions may allow investigators to study mechanotransduction in physiologically relevant environments. These platforms could provide valuable experimental bridges between molecular studies and in vivo cardiovascular physiology.
Despite substantial progress, several major challenges remain. Cardiovascular mechanotransduction involves highly dynamic and nonlinear interactions across vastly different temporal and spatial scales, making computational integration difficult. Mechanical forces continuously evolve during disease progression, and mechanosensitive responses are strongly influenced by cell type, tissue architecture, and biomechanical context. Developing predictive multiscale models, therefore, requires extensive interdisciplinary collaboration among mechanobiologists, engineers, computational scientists, physiologists, and clinicians.
Overall, multiscale cardiovascular mechanobiology represents a rapidly evolving field with enormous potential for advancing understanding of cardiovascular disease mechanisms. Integrating molecular mechanosensing, cellular signaling, tissue biomechanics, organ-level physiology, computational modeling, and systems biology will be essential for elucidating how mechanical forces regulate cardiovascular adaptation and pathology. Continued development of multiscale approaches is expected to significantly improve mechanistic understanding, therapeutic development, and precision medicine strategies targeting PIEZO-mediated cardiovascular mechanotransduction.
6.3. Precision Medicine Applications
Precision medicine has emerged as a transformative approach in cardiovascular healthcare by emphasizing individualized diagnosis, risk prediction, and therapeutic intervention based on genetic, molecular, physiological, and environmental variability. Rather than relying solely on generalized treatment strategies, precision medicine seeks to tailor therapies according to patient-specific disease mechanisms and biological characteristics. As understanding of cardiovascular mechanobiology expands, mechanotransduction pathways—particularly those involving PIEZO channels—are increasingly recognized as potential contributors to interindividual variability in cardiovascular disease susceptibility and therapeutic responsiveness [51,56]. Consequently, targeting PIEZO-mediated signaling pathways may eventually enable highly personalized cardiovascular therapies based on mechanobiological phenotypes and genetic profiles.
Genetic variability in PIEZO channels has already been linked to several human diseases, demonstrating the profound physiological importance of mechanosensitive ion channel regulation. Mutations in PIEZO1 are associated with hereditary xerocytosis, altered erythrocyte hydration, lymphatic abnormalities, and other mechanotransduction-related disorders [13]. These findings suggest that naturally occurring variations in PIEZO channel structure, gating kinetics, or expression levels may significantly influence cardiovascular physiology and disease risk. In the cardiovascular system, such genetic differences may alter endothelial shear stress sensing, calcium homeostasis, inflammatory signaling, fibrosis, and myocardial adaptation to mechanical stress.
Emerging evidence suggests that individual differences in PIEZO-mediated mechanotransduction may influence susceptibility to hypertension, cardiac hypertrophy, fibrosis, ischemic injury, and vascular inflammation [26,28]. For example, variations that enhance PIEZO1 mechanosensitivity could potentially increase calcium influx and inflammatory activation under chronic hemodynamic stress, thereby accelerating pathological remodeling and cardiovascular disease progression. Conversely, reduced mechanotransduction sensitivity may impair adaptive vascular responses to shear stress and contribute to endothelial dysfunction or abnormal cardiovascular regulation.
Precision medicine approaches targeting mechanotransduction pathways may therefore help identify patients with increased vulnerability to biomechanical cardiovascular stress. Integration of genomic screening with biomechanical and clinical assessment could potentially reveal mechanotransduction-related risk profiles associated with hypertension, heart failure, arrhythmias, or vascular disease. Such strategies may be especially valuable in diseases strongly influenced by mechanical loading conditions, including hypertensive heart disease, valvular disease, cardiomyopathy, and atherosclerosis.
One important future direction involves the development of personalized mechanobiological phenotyping. In addition to genetic variability, mechanotransduction responses are influenced by extracellular matrix composition, vascular stiffness, cytoskeletal organization, membrane lipid composition, and hemodynamic conditions [62]. Advanced imaging technologies, hemodynamic monitoring, and biomechanical modeling may allow patient-specific assessment of vascular stiffness, myocardial strain, shear stress distribution, and tissue remodeling patterns. Combining these biomechanical parameters with genomic and molecular data could provide highly individualized mechanobiological profiles for cardiovascular risk stratification and therapeutic planning.
Artificial intelligence and machine learning are expected to play increasingly important roles in precision mechanobiology. AI-driven computational frameworks can integrate genomic data, electrophysiology, imaging, biomechanics, proteomics, and clinical outcomes to build predictive models that identify patient-specific mechanotransduction abnormalities [59]. Machine learning algorithms may help detect hidden relationships between PIEZO-related genetic variants and cardiovascular phenotypes, thereby improving the prediction of disease progression and treatment responsiveness.
Personalized mechanotransduction analysis may also improve therapeutic targeting. Because PIEZO signaling contributes to multiple cardiovascular processes—including endothelial regulation, fibroblast activation, hypertrophic remodeling, inflammation, and calcium homeostasis—patients may exhibit different pathological mechanisms depending on their genetic and biomechanical backgrounds. Precision medicine approaches could therefore help identify individuals most likely to benefit from mechanotransduction-targeted therapies such as PIEZO inhibitors, anti-fibrotic agents, anti-inflammatory therapies, or calcium signaling modulators [72].
For example, patients with heightened PIEZO-mediated inflammatory activation may particularly benefit from therapies targeting endothelial mechanotransduction or inflammatory signaling pathways. Similarly, individuals exhibiting enhanced mechanosensitive hypertrophic signaling could potentially respond more effectively to therapies aimed at reducing calcium overload, calcineurin/NFAT activation, or mechanosensitive remodeling pathways [39]. Personalized treatment selection based on mechanobiological phenotypes may therefore improve therapeutic efficacy while reducing adverse effects.
Precision medicine approaches may also support personalized prevention strategies. Early identification of individuals genetically predisposed to abnormal mechanotransduction could facilitate targeted interventions before the development of irreversible cardiovascular remodeling. Lifestyle modifications, blood pressure control, exercise recommendations, and pharmacological therapies might eventually be tailored according to patient-specific mechanobiological risk factors.
Another promising future application involves integrating patient-specific computational models, or “digital twins”, into cardiovascular medicine. Digital twin technologies combine imaging, hemodynamics, molecular data, and computational biomechanics to create individualized virtual models of cardiovascular function. Incorporating PIEZO-mediated mechanotransduction into these models could allow simulation of disease progression, mechanical stress responses, and therapeutic outcomes under different clinical scenarios. Such predictive platforms may eventually support individualized treatment planning and precision cardiovascular care.
Advances in genomic sequencing technologies are also likely to accelerate precision mechanotransduction research. Large-scale population studies may identify novel PIEZO variants associated with cardiovascular diseases, vascular remodeling, or differential therapeutic responsiveness. Understanding how specific genetic variants influence channel structure, mechanosensitivity, gating kinetics, and downstream signaling will be essential for translating mechanobiological discoveries into precision therapies.
In addition to genomics, epigenetic and epitranscriptomic regulation may contribute substantially to interindividual differences in mechanotransduction. Emerging studies suggest that mechanical stress can influence RNA modification, chromatin organization, and transcriptional regulation of fibrotic and inflammatory pathways [81]. Personalized assessment of these regulatory mechanisms may further refine mechanobiological risk prediction and therapeutic targeting.
Despite these exciting possibilities, several major challenges remain before precision mechanotransduction medicine can be fully realized. PIEZO channels exhibit widespread physiological functions throughout multiple organ systems, making selective therapeutic targeting difficult. Furthermore, cardiovascular mechanotransduction involves highly dynamic interactions among genetics, biomechanics, metabolism, inflammation, and environmental factors. Developing clinically useful predictive models will therefore require extensive integration of molecular biology, biomechanics, computational modeling, and longitudinal clinical data.
Another challenge involves the complexity of mechanobiological measurements. Accurate assessment of tissue stiffness, shear stress, membrane mechanics, and cellular force transmission in clinical settings remains technically demanding. Standardized biomarkers and imaging methods for evaluating mechanotransduction activity will likely be necessary for widespread clinical implementation.
Overall, precision medicine applications targeting PIEZO-mediated mechanotransduction represent a highly promising future direction in cardiovascular research and therapy. Genetic variability in PIEZO channels and mechanotransduction pathways may significantly influence cardiovascular disease susceptibility, remodeling patterns, and therapeutic responsiveness. By integrating genomics, biomechanics, computational modeling, artificial intelligence, and clinical phenotyping, future precision medicine approaches may enable highly personalized cardiovascular therapies targeting pathological mechanical signaling while preserving essential physiological mechanotransduction functions.
6.4. Future Challenges and Perspectives
Despite major advances in understanding PIEZO-mediated mechanotransduction, numerous scientific, technological, and translational challenges remain unresolved. Cardiovascular mechanobiology is inherently complex because mechanical forces influence biological systems across multiple spatial and temporal scales, ranging from nanoscale membrane deformation to organ-level hemodynamic remodeling. Although substantial progress has been achieved in structural biology, electrophysiology, molecular genetics, and computational modeling, many fundamental aspects of PIEZO channel regulation and cardiovascular function remain incompletely understood. Addressing these challenges will be essential for translating mechanobiological discoveries into clinically effective therapies.
One of the most important unresolved questions involves the precise molecular mechanisms governing PIEZO channel gating and force transmission. Increasing evidence suggests that cytoskeletal interactions, extracellular matrix mechanics, membrane lipid composition, and intracellular scaffolding proteins substantially influence mechanosensitivity and gating behavior [14,21,62]. The relative contributions of these regulatory mechanisms likely vary among different cardiovascular cell types and physiological conditions. Further investigation is therefore needed to clarify how mechanical forces are transmitted from the extracellular environment to PIEZO channels and how these pathways are altered during disease progression.
Another major challenge involves distinguishing the specific physiological and pathological roles of PIEZO1 and PIEZO2 within the cardiovascular system. While PIEZO1 has emerged as the dominant cardiovascular mechanosensor in endothelial cells, cardiomyocytes, fibroblasts, and erythrocytes, the functions of PIEZO2 in cardiovascular biology remain comparatively poorly understood. Some studies suggest overlapping mechanosensory functions, whereas others indicate distinct tissue-specific roles and regulatory mechanisms [25,56]. Clarifying the differential contributions of PIEZO isoforms will be critical for understanding mechanotransduction specificity and for developing targeted therapeutic strategies.
A further challenge arises from the highly context-dependent nature of mechanotransduction signaling. Mechanical responses are strongly influenced by local hemodynamic conditions, extracellular matrix stiffness, membrane composition, inflammatory state, metabolic regulation, and cell-cell interactions. Consequently, PIEZO-mediated signaling may produce protective adaptive responses under physiological conditions while promoting pathological remodeling under chronic or excessive mechanical stress. For example, moderate PIEZO activation may support vascular adaptation and cardiac performance, whereas sustained activation may contribute to calcium overload, fibrosis, inflammation, and progression of heart failure [27,39]. Understanding these context-dependent mechanobiological responses remains essential for therapeutic development.
Translational targeting of PIEZO channels also presents substantial pharmacological challenges. Because PIEZO channels are widely expressed throughout multiple organ systems and participate in diverse physiological processes, systemic inhibition or activation may produce unintended off-target effects. Current pharmacological modulators of PIEZO channels remain relatively limited in specificity and mechanistic selectivity [72]. Furthermore, tissue-specific differences in mechanotransduction signaling complicate therapeutic design. Development of selective modulators capable of targeting pathological mechanotransduction while preserving normal physiological function, therefore, remains a major priority in the field.
Another important limitation involves current experimental models used to study cardiovascular mechanotransduction. Many mechanobiological investigations rely on simplified in vitro systems that may not fully reproduce the highly dynamic biomechanical environments present in living cardiovascular tissues. Traditional two-dimensional cell culture models often fail to replicate physiological extracellular matrix architecture, hemodynamic loading, and multicellular interactions. Similarly, animal models may not completely capture the complexity of human cardiovascular disease progression. Advanced experimental systems, including organ-on-chip technologies, engineered cardiac tissues, three-dimensional vascular models, and patient-specific induced pluripotent stem cell platforms, may help overcome some of these limitations by providing more physiologically relevant mechanobiological environments.
Artificial intelligence and machine learning approaches offer enormous promise for mechanotransduction research but also introduce important computational and methodological challenges. Deep learning models require large, high-quality datasets for training and validation, yet electrophysiological recordings of mechanosensitive ion channels are often noisy, heterogeneous, and difficult to obtain experimentally. Overfitting, limited generalizability, and lack of interpretability remain important concerns in AI-assisted ion channel analysis. In many cases, neural network models function as “black boxes”, making it difficult to determine which electrophysiological features drive predictions or hidden-state classifications. Future development of explainable artificial intelligence frameworks and standardized electrophysiological datasets will therefore be important for improving reliability and translational utility.
Multiscale integration represents another major frontier in cardiovascular mechanobiology. Mechanical forces influence biological systems across molecular, cellular, tissue, organ, and systemic scales, yet integrating these scales into unified mechanistic frameworks remains highly challenging. Molecular conformational changes within PIEZO channels ultimately influence tissue remodeling, vascular stiffness, myocardial mechanics, and organ-level cardiovascular function. Developing computational and experimental frameworks capable of linking nanoscale mechanosensing to whole-organ physiology will require extensive interdisciplinary collaboration among mechanobiologists, engineers, computational scientists, and clinicians.
Patient-specific variability also presents an important challenge for future mechanotransduction research. Genetic differences, age-related changes, sex-specific physiology, metabolic conditions, and environmental factors may all influence PIEZO channel expression, mechanosensitivity, and downstream signaling pathways. Emerging evidence suggests that patient-specific mechanobiological phenotypes may contribute to differential susceptibility to hypertension, fibrosis, arrhythmias, and heart failure. Precision medicine approaches integrating genomics, biomechanics, computational modeling, and clinical phenotyping may therefore become increasingly important for individualized cardiovascular therapy.
In addition, the interactions between mechanotransduction and other signaling systems remain incompletely understood. PIEZO-mediated calcium signaling interfaces with inflammatory pathways, oxidative stress responses, metabolic regulation, epigenetic remodeling, and neurohumoral activation. Emerging evidence suggests that mechanobiology may influence chromatin organization, RNA modification, and gene expression networks involved in fibrosis and cardiovascular disease progression [74]. Understanding these complex signaling interactions will be essential for developing comprehensive mechanobiological models of cardiovascular pathology.
Future therapeutic development may also require combination strategies targeting both mechanical signaling and downstream pathological pathways. Because cardiovascular diseases such as hypertension, fibrosis, and heart failure involve highly interconnected molecular networks, modulation of PIEZO channels alone may be insufficient to fully reverse disease progression. Integrating mechanotransduction-targeted therapies with anti-inflammatory, anti-fibrotic, metabolic, or regenerative interventions may ultimately prove more effective.
Despite these challenges, the future of cardiovascular mechanobiology remains highly promising. Rapid advances in cryo-electron microscopy, high-resolution electrophysiology, molecular dynamics simulations, artificial intelligence, multiscale computational modeling, and precision medicine are providing unprecedented insights into the molecular basis of mechanotransduction. Continued interdisciplinary research is expected to significantly improve understanding of PIEZO channel regulation, cardiovascular adaptation, and disease progression.
Despite substantial advances in cardiovascular mechanobiology, many important questions regarding PIEZO-mediated signaling remain unresolved. The integration of PIEZO channels with broader mechanosensory networks involving integrins, focal adhesions, TRP channels, cytoskeletal force transmission, extracellular matrix mechanics, and YAP/TAZ signaling requires further investigation [5,6,7]. In addition, most current evidence derives from experimental cellular and rodent studies, emphasizing the need for more comprehensive human mechanobiological datasets and patient-specific translational investigations.
Emerging computational approaches, including artificial intelligence-assisted electrophysiology, multiscale biomechanical simulations, and machine learning-assisted drug discovery, may substantially accelerate understanding of force-dependent cardiovascular signaling and facilitate the development of precision mechanobiology approaches for cardiovascular medicine.
Overall, PIEZO-mediated mechanotransduction represents one of the most important emerging frontiers in cardiovascular science. Although substantial challenges remain, ongoing advances in structural biology, biomechanics, computational modeling, and translational medicine are steadily transforming mechanobiological research from a primarily descriptive field into a mechanistically integrated and clinically relevant discipline. Future investigation of PIEZO signaling pathways may ultimately lead to novel diagnostic tools, precision mechanomedicine approaches, and targeted therapies capable of preventing or reversing mechanically driven cardiovascular disease.
7. Conclusions
Mechanical forces are fundamental regulators of cardiovascular physiology and disease, and PIEZO channels have emerged as central mediators of cardiovascular mechanotransduction. Over the past decade, major advances in structural biology, electrophysiology, molecular genetics, and computational modeling have substantially improved understanding of how PIEZO channels convert mechanical stimuli into intracellular biochemical and electrophysiological responses. PIEZO-mediated signaling regulates a wide range of cardiovascular processes, including endothelial shear stress sensing, vascular tone regulation, cardiac remodeling, fibroblast activation, inflammatory signaling, and erythrocyte deformability.
Accumulating evidence demonstrates that dysregulated PIEZO signaling contributes to numerous cardiovascular disorders, including hypertension, cardiac hypertrophy, fibrosis, vascular inflammation, ischemic injury, and heart failure progression. These findings highlight the importance of mechanotransduction not only as a fundamental physiological process but also as a major driver of cardiovascular pathology. At the same time, the widespread involvement of PIEZO channels in cardiovascular adaptation and disease identifies mechanotransduction pathways as promising therapeutic targets.
Recent integration of artificial intelligence, machine learning, and multiscale computational modeling is further transforming mechanobiology research by enabling improved analysis of electrophysiological data, ion channel kinetics, and force-dependent signaling dynamics. These emerging approaches may accelerate the development of precision mechanomedicine strategies and novel therapeutics targeting mechanically driven cardiovascular disease.
Despite substantial progress, many important challenges remain unresolved, including tissue-specific regulation of PIEZO signaling, integration of multiscale mechanotransduction processes, selective pharmacological targeting, and translation of mechanobiological discoveries into clinical therapies. Continued interdisciplinary collaboration among biologists, engineers, computational scientists, and clinicians will therefore be essential for advancing the field.
Overall, PIEZO-mediated mechanotransduction represents one of the most rapidly evolving frontiers in cardiovascular science. Future investigation of mechanosensitive signaling pathways is expected to provide deeper insight into cardiovascular adaptation, disease progression, and therapeutic intervention, ultimately contributing to the development of innovative strategies to prevent and treat cardiovascular disease.
Statement of the Use of Generative AI and AI-Assisted Technologies in the Writing Process
During the preparation of this manuscript/study, the authors used ChatGPT 5.2 for the purposes of grammar, editing, conceptual figure preparation, and structural refinement of the manuscript. The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were generated or analyzed in this study. All data supporting the findings of this review are derived from previously published sources cited in the manuscript.
Funding
This research received no external funding.
Declaration of Competing Interest
The author declares that he has no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
Davies PF. Flow-mediated endothelial mechanotransduction. Physiol. Rev. 1995, 75, 519–560. DOI:10.1152/physrev.1995.75.3.519 [Google Scholar]
-
Davis MJ, Earley S, Li YS, Chien S. Vascular Mechanotransduction. Physiol. Rev. 2023, 103, 1247–1421. DOI:10.1152/physrev.00053.2021 [Google Scholar]
-
Saucerman JJ, Tan PM, Buchholz KS, McCulloch AD, Omens JH. Mechanical regulation of gene expression in cardiac myocytes and fibroblasts. Nat. Rev. Cardiol. 2019, 16, 361–378. DOI:10.1038/s41569-019-0155-8 [Google Scholar]
-
Wang Y, Chatterjee E, Li G, Xu J, Xiao J. Force-sensing protein expression in response to cardiovascular mechanotransduction. eBioMedicine 2024, 110, 105412. DOI:10.1016/j.ebiom.2024.105412 [Google Scholar]
-
Hahn C, Schwartz MA. Mechanotransduction in vascular physiology and atherogenesis. Nat. Rev. Mol. Cell Biol 2009, 10, 53–62. DOI:10.1038/nrm2596 [Google Scholar]
-
Ingber DE. Mechanobiology and diseases of mechanotransduction. Ann. Med. 2003, 35, 564–577. DOI:10.1080/07853890310016333 [Google Scholar]
-
Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. DOI:10.1038/nature10137 [Google Scholar]
-
Earley S, Brayden JE. Transient receptor potential channels and vascular function. Clin. Sci. 2010, 119, 19–36. DOI:10.1042/Cs20090641 [Google Scholar]
-
Kuwahara K, Wang YG, McAnally J, Richardson JA, Bassel-Duby R, Hill JA, et al. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J. Clin. Investig. 2006, 116, 3114–3126. DOI:10.1172/jci27702 [Google Scholar]
-
Köhler R, Heyken WT, Heinau P, Schubert R, Si H, Kacik M, et al. Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1495–1502. DOI:10.1161/01.ATV.0000225698.36212.6a [Google Scholar]
-
Martinac B, Poole K. Mechanically activated ion channels. Int. J. Biochem. Cell Biol. 2018, 97, 104–107. DOI:10.1016/j.biocel.2018.02.011 [Google Scholar]
-
Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, et al. Piezo1 and Piezo2 Are Essential Components of Distinct Mechanically Activated Cation Channels. Science 2010, 330, 55–60. DOI:10.1126/science.1193270 [Google Scholar]
-
Syeda R. Physiology and Pathophysiology of Mechanically Activated PIEZO Channels. Annu. Rev. Neurosci. 2021, 44, 383–402. DOI:10.1146/annurev-neuro-093020-120939 [Google Scholar]
-
Cox CD, Bavi N, Martinac B. Origin of the Force: The Force-From-Lipids Principle Applied to Piezo Channels. Curr. Top. Membr. 2017, 79, 59–96. DOI:10.1016/bs.ctm.2016.09.001 [Google Scholar]
-
Ge J, Li W, Zhao Q, Li N, Chen M, Zhi P, et al. Architecture of the mammalian mechanosensitive Piezo1 channel. Nature 2015, 527, 64–69. DOI:10.1038/nature15247 [Google Scholar]
-
Saotome K, Murthy SE, Kefauver JM, Whitwam T, Patapoutian A, Ward AB. Structure of the mechanically activated ion channel Piezol. Nature 2018, 554, 481–486. DOI:10.1038/nature25453 [Google Scholar]
-
Zhao Q, Zhou H, Chi S, Wang Y, Wang J, Geng J, et al. Structure and mechanogating mechanism of the Piezo1 channel. Nature 2018, 554, 487–492. DOI:10.1038/nature25743 [Google Scholar]
-
Lin YC, Guo YSR, Miyagi A, Levring J, MacKinnon R, Scheuring S. Structural Response of the Piezo Channel Upon Application of Force. Biophys. J. 2019, 116, 301a. DOI:DOI 10.1016/j.bpj.2018.11.1633 [Google Scholar]
-
Young M, Lewis AH, Grandl J. Physics of mechanotransduction by Piezo ion channels. J. Gen. Physiol. 2022, 154, e202113044. DOI:10.1085/jgp.202113044 [Google Scholar]
-
Lewis AH, Grandl J. Mechanical sensitivity of Piezo1 ion channels can be tuned by cellular membrane tension. Elife 2015, 4, e12088. DOI:10.7554/eLife.12088 [Google Scholar]
-
Cox CD, Bae C, Ziegler L, Hartley S, Nikolova-Krstevski V, Rohde PR, et al. Removal of the mechanoprotective influence of the cytoskeleton reveals PIEZO1 is gated by bilayer tension. Nat. Commun. 2016, 7, 10366. DOI:10.1038/ncomms10366 [Google Scholar]
-
Moroni M, Servin-Vences MR, Fleischer R, Sánchez-Carranza O, Lewin GR. Voltage gating of mechanosensitive PIEZO channels. Nat. Commun. 2018, 9, 1096. DOI:10.1038/s41467-018-03502-7 [Google Scholar]
-
Verkest C, Lechner SG. Advances and recent insights into the gating mechanisms of the mechanically activated ion channels PIEZO1 and PIEZO2. Curr. Opin. Physiol. 2023, 31, 100625. DOI:10.1016/j.cophys.2022.100625 [Google Scholar]
-
Ranade SS, Woo SH, Dubin AE, Moshourab RA, Wetzel C, Petrus M, et al. Piezo2 is the major transducer of mechanical forces for touch sensation in mice. Nature 2014, 516, 121–125. DOI:10.1038/nature13980 [Google Scholar]
-
Chesler AT, Szczot M, Bharucha-Goebel D, Ceko M, Donkervoort S, Laubacher C, et al. The Role of PIEZO2 in Human Mechanosensation. N. Engl. J. Med. 2016, 375, 1355–1364. DOI:10.1056/NEJMoa1602812 [Google Scholar]
-
Beech DJ, Kalli AC. Force Sensing by Piezo Channels in Cardiovascular Health and Disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2228–2239. DOI:10.1161/atvbaha.119.313348 [Google Scholar]
-
Douguet D, Patel A, Xu AM, Vanhoutte PM, Honoré E. Piezo Ion Channels in Cardiovascular Mechanobiology. Trends Pharmacol. Sci. 2019, 40, 956–970. DOI:10.1016/j.tips.2019.10.002 [Google Scholar]
-
Coste B, Delmas P. PIEZO Ion Channels in Cardiovascular Functions and Diseases. Circ. Res. 2024, 134, 572–591. DOI:10.1161/Circresaha.123.322798 [Google Scholar]
-
Li J, Hou B, Tumova S, Muraki K, Bruns A, Ludlow MJ, et al. Piezo1 integration of vascular architecture with physiological force. Nature 2014, 515, 279–282. DOI:10.1038/nature13701 [Google Scholar]
-
Wang S, Chennupati R, Kaur H, Iring A, Wettschureck N, Offermanns S. Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J. Clin. Investig. 2016, 126, 4527–4536. DOI:10.1172/JCI87343 [Google Scholar]
-
Jiang F, Yin K, Wu K, Zhang M, Wang S, Cheng H, et al. The mechanosensitive Piezo1 channel mediates heart mechano-chemo transduction. Nat. Commun. 2021, 12, 869. DOI:10.1038/s41467-021-21178-4 [Google Scholar]
-
Jiang Y, Yang XZ, Jiang JH, Xiao BL. Structural Designs and Mechanogating Mechanisms of the Mechanosensitive Piezo Channels. Trends Biochem. Sci. 2021, 46, 472–488. DOI:10.1016/j.tibs.2021.01.008 [Google Scholar]
-
Liang J, Huang B, Yuan G, Chen Y, Liang F, Zeng H, et al. Stretch-activated channel Piezo1 is up-regulated in failure heart and cardiomyocyte stimulated by AngII. Am. J. Transl. Res. 2017, 9, 2945–2955. Available online: https://pmc.ncbi.nlm.nih.gov/articles/PMC5489894/ (accessed on 5 May 2026).
-
Wong TY, Juang WC, Tsai CT, Tseng CJ, Lee WH, Chang SN, et al. Mechanical Stretching Simulates Cardiac Physiology and Pathology through Mechanosensor Piezo1. J. Clin. Med. 2018, 7, 410. DOI:10.3390/jcm7110410 [Google Scholar]
-
Yu ZY, Gong H, Kesteven S, Guo Y, Wu J, Li JV, et al. Piezo1 is the cardiac mechanosensor that initiates the cardiomyocyte hypertrophic response to pressure overload in adult mice. Nat. Cardiovasc. Res. 2022, 1, 577–591. DOI:10.1038/s44161-022-00082-0 [Google Scholar]
-
Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair 2012, 5, 15. DOI:10.1186/1755-1536-5-15 [Google Scholar]
-
Blythe NM, Muraki K, Ludlow MJ, Stylianidis V, Gilbert HTJ, Evans EL, et al. Mechanically activated Piezo1 channels of cardiac fibroblasts stimulate p38 mitogen-activated protein kinase activity and interleukin-6 secretion. J. Biol. Chem. 2019, 294, 17395–17408. DOI:10.1074/jbc.RA119.009167 [Google Scholar]
-
Emig R, Knodt W, Krussig MJ, Zgierski-Johnston CM, Gorka O, Gross O, et al. Piezo1 Channels Contribute to the Regulation of Human Atrial Fibroblast Mechanical Properties and Matrix Stiffness Sensing. Cells 2021, 10, 663. DOI:10.3390/cells10030663 [Google Scholar]
-
Zhang Y, Su SA, Li W, Ma Y, Shen J, Wang Y, et al. Piezo1-Mediated Mechanotransduction Promotes Cardiac Hypertrophy by Impairing Calcium Homeostasis to Activate Calpain/Calcineurin Signaling. Hypertension 2021, 78, 647–660. DOI:10.1161/HYPERTENSIONAHA.121.17177 [Google Scholar]
-
Umbarkar P, Ramirez SR, Ejantkar S, Tousif S, Cora AT, Galindo CL, et al. Mechanosensitive Piezo-1 Channel Aggravates Ischemia Induced Adverse Cardiac Remodeling and Dysfunction. Circ. Res. 2023, 133 (Suppl. S1), AP2005. DOI:10.1161/res.133.suppl_1.P2005 [Google Scholar]
-
Nagase T, Nagase M. Piezo ion channels: Long-sought-after mechanosensors mediating hypertension and hypertensive nephropathy. Hypertens. Res. 2024, 47, 2786–2799. DOI:10.1038/s41440-024-01820-6 [Google Scholar]
-
Baratchi S, Zaldivia MTK, Wallert M, Loseff-Silver J, Al-Aryahi S, Zamani J, et al. Transcatheter Aortic Valve Implantation Represents an Anti-Inflammatory Therapy Via Reduction of Shear Stress-Induced, Piezo-1-Mediated Monocyte Activation. Circulation 2020, 142, 1092–1105. DOI:10.1161/Circulationaha.120.045536 [Google Scholar]
-
Choi D, Park E, Yu RP, Cooper MN, Cho IT, Choi J, et al. Piezo1-Regulated Mechanotransduction Controls Flow-Activated Lymphatic Expansion. Circ. Res. 2022, 131, e2–e21. DOI:10.1161/CIRCRESAHA.121.320565 [Google Scholar]
-
Zhao FJ, Li FF, Sedaghati F, Dong H, Kumar S, Foster DG, et al. Piezo-type mechanosensitive ion channel component 1 (PIEZO1) is upregulated in peripheral arterial disease (PAD) and a novel murine PAD model. JVS-Vasc. Sci. 2025, 6, 100394. DOI:10.1016/j.jvssci.2025.100394 [Google Scholar]
-
Xiao R, Xu XZS. Mechanosensitive Channels: In Touch with Piezo. Curr. Biol. 2010, 20, R936–R938. DOI:10.1016/j.cub.2010.09.053 [Google Scholar]
-
Coste B. Piezo proteins form a new class of mechanically activated ion channels. Med. Sci. 2012, 28, 1056–1057. DOI:10.1051/medsci/20122812012 [Google Scholar]
-
Coste B, Xiao BL, Santos JS, Syeda R, Grandl J, Spencer KS, et al. Piezo proteins are pore-forming subunits of mechanically activated channels. Nature 2012, 483, 176–181. DOI:10.1038/nature10812 [Google Scholar]
-
Volkers L, Mechioukhi Y, Coste B. Piezo channels: from structure to function. Pflug. Arch. Eur. J. Physiol. 2015, 467, 95–99. DOI:10.1007/s00424-014-1578-z [Google Scholar]
-
Geng J, Zhao Q, Zhang T, Xiao B. In Touch With the Mechanosensitive Piezo Channels: Structure, Ion Permeation, and Mechanotransduction. Curr. Top. Membr. 2017, 79, 159–195. DOI:10.1016/bs.ctm.2016.11.006 [Google Scholar]
-
Lin YC, Guo YR, Miyagi A, Levring J, MacKinnon R, Scheuring S. Force-induced conformational changes in PIEZO1. Nature 2019, 573, 230–234. DOI:10.1038/s41586-019-1499-2 [Google Scholar]
-
Xiao BL. Mechanisms of mechanotransduction and physiological roles of PIEZO channels. Nat. Rev. Mol. Cell Biol. 2024, 25, 886–903. DOI:10.1038/s41580-024-00773-5 [Google Scholar]
-
Xiao BL. Structural and Functional Characterizations of the Mechanosensitive Piezo Channel. Biophys. J. 2016, 110, 349a. DOI:10.1016/j.bpj.2015.11.1879 [Google Scholar]
-
Liu WH, Zhang XC, He QJ, Meng Y, Tian BX, Xiao BL. Spring-like mechanics enable rapid inactivation and stochastic single-channel gating of the mechanically activated PIEZO channel. Cell Rep. 2025, 44, 116615. DOI:10.1016/j.celrep.2025.116615 [Google Scholar]
-
Zhao QC, Wu K, Geng J, Chi SP, Wang YF, Zhi P, et al. Ion Permeation and Mechanotransduction Mechanisms of Mechanosensitive Piezo Channels. Neuron 2016, 89, 1248–1263. DOI:10.1016/j.neuron.2016.01.046 [Google Scholar]
-
Gnanasambandam R, Gottlieb PA, Sachs F. The Kinetics and the Permeation Properties of Piezo Channels. Curr. Top. Membr. 2017, 79, 275–307. DOI:10.1016/bs.ctm.2016.11.004 [Google Scholar]
-
Murthy SE, Dubin AE, Patapoutian A. Piezos thrive under pressure: mechanically activated ion channels in health and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 771–783. DOI:10.1038/nrm.2017.92 [Google Scholar]
-
Wijerathne TD, Lacroix JJ. Kinetics and permeation properties of mammalian Piezo channels. Biophys. J. 2024, 123, 331a–332a. DOI:10.1016/j.bpj.2023.11.2021 [Google Scholar]
-
Wu J, Lewis AH, Grandl J. Touch, Tension, and Transduction—The Function and Regulation of Piezo Ion Channels. Trends Biochem. Sci. 2017, 42, 57–71. DOI:10.1016/j.tibs.2016.09.004 [Google Scholar]
-
Fang XZ, Zhou T, Xu JQ, Wang YX, Sun MM, He YJ, et al. Structure, kinetic properties and biological function of mechanosensitive Piezo channels. Cell Biosci. 2021, 11, 13. DOI:10.1186/s13578-020-00522-z [Google Scholar]
-
Syeda R, Florendo MN, Cox CD, Kefauver JM, Santos JS, Martinac B, et al. Piezo1 Channels Are Inherently Mechanosensitive. Cell Rep. 2016, 17, 1739–1746. DOI:10.1016/j.celrep.2016.10.033 [Google Scholar]
-
Cox CD. PIEZO channel auxiliary subunits in sensory and non-sensory cells. Biophys. J. 2026, 125, 3a. DOI:10.1016/j.bpj.2025.11.231 [Google Scholar]
-
Richardson J, Kotevski A, Poole K. From stretch to deflection: the importance of context in the activation of mammalian, mechanically activated ion channels. FEBS J. 2022, 289, 4447–4469. DOI:10.1111/febs.16041 [Google Scholar]
-
Borbiro I, Rohacs T. Regulation of Piezo Channels by Cellular Signaling Pathways. Curr. Top. Membr. 2017, 79, 245–261. DOI:10.1016/bs.ctm.2016.10.002 [Google Scholar]
-
Wijerathne TD, Bhatt A, Jiang WJ, Luo YL, Lacroix JJ. Mammalian PIEZO channels rectify anionic currents. Biophys. J. 2025, 124, 4424–4431. DOI:10.1016/j.bpj.2024.11.010 [Google Scholar]
-
Lewis AH, Grandl J. Stretch and poke stimulation for characterizing mechanically activated ion channels. Methods Enzymol. 2021, 654, 225–253. DOI:10.1016/bs.mie.2020.12.024 [Google Scholar]
-
Gottlieb PA. The Discovery, Properties, and Function of Piezo Channels. Curr. Top. Membr. 2017, 79, 1–36. DOI:10.1016/bs.ctm.2016.11.007 [Google Scholar]
-
Grandl J. The Mechanism of Activation of Piezo Ion Channels. Biophys. J. 2017, 112, 37a–38a. DOI:10.1016/j.bpj.2016.11.237 [Google Scholar]
-
Celik N, O’Brien F, Brennan S, Rainbow RD, Dart C, Zheng YL, et al. Deep-Channel uses deep neural networks to detect single-molecule events from patch-clamp data. Commun. Biol. 2020, 3, 3. DOI:10.1038/s42003-019-0729-3 [Google Scholar]
-
Haselwandter C. Force-sensing in Piezo ion channels. Biophys. J. 2023, 122, 159a–160a. DOI:10.1016/j.bpj.2022.11.953 [Google Scholar]
-
Ray A, Haselwandter CA. Mechanics of Force Sensing in Piezo Ion Channels. Eur. Biophys. J. Biophys. 2025, 54, S163. DOI:10.1007/s00249-025-01757-9 [Google Scholar]
-
Liu YS, Liu BW, Zhan WR, Zhang L, Shi AB, Lin L, et al. Membrane dome-based regulation mechanism of the mechanosensitive PIEZO channel. Cell Rep. 2025, 44, 116651. DOI:10.1016/j.celrep.2025.116651 [Google Scholar]
-
Kinsella JA, Debant M, Parsonage G, Morley LC, Bajarwan M, Revill C, et al. Pharmacology of PIEZO1 channels. Br. J. Pharmacol. 2024, 181, 4714–4732. DOI:10.1111/bph.17351 [Google Scholar]
-
Beech DJ, Fagnen C, Kalli AC. Biological sensing of fluid flow-lessons from PIEZO1. Biophys. Rev. 2024, 16, 871–873. DOI:10.1007/s12551-024-01246-x [Google Scholar]
-
Rode B, Shi J, Endesh N, Drinkhill MJ, Webster PJ, Lotteau SJ, et al. Piezo1 channels sense whole body physical activity to reset cardiovascular homeostasis and enhance performance. Nat. Commun. 2017, 8, 350. DOI:10.1038/s41467-017-00429-3 [Google Scholar]
-
Ranade SS, Qiu Z, Woo SH, Hur SS, Murthy SE, Cahalan SM, et al. Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10347–10352. DOI:10.1073/pnas.1409233111 [Google Scholar]
-
Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. DOI:10.1038/s41569-018-0007-y [Google Scholar]
-
McGrane A, Murray M, Bartoli F, Giannoudi M, Conning-Rowland M, Stewart L, et al. PIEZO Force Sensors and the Heart. Cold Spring Harb. Perspect. Biol. 2026, 18, a041806. DOI:10.1101/cshperspect.a041806 [Google Scholar]
-
Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J. Mol. Cell Cardiol. 2011, 51, 468–473. DOI:10.1016/j.yjmcc.2011.01.012 [Google Scholar]
-
Murphy JF, Li X. The roles of the microtubule cytoskeletal network in cardiac mechanobiology. J. Cell Sci. 2026, 139, jcs264308. DOI:10.1242/jcs.264308 [Google Scholar]
-
Chen Q, Zhang H, Chen Y, Peng Y, Yao Y, Xue H, et al. Trimethylamine N-oxide induces cardiac diastolic dysfunction by down-regulating Piezo1 in mice with heart failure with preserved ejection fraction. Life Sci. 2025, 369, 123554. DOI:10.1016/j.lfs.2025.123554 [Google Scholar]
-
Garoffolo G, Pesce M. When cell mechanics meets epitranscriptomics: reduction of m6A in Piezo2 RNA ameliorates cardiac fibrosis. Cardiovasc. Res. 2024, 120, 2158–2159. DOI:10.1093/cvr/cvae247 [Google Scholar]
-
Huisjes R, Bogdanova A, van Solinge WW, Schiffelers RM, Kaestner L, van Wijk R. Squeezing for Life—Properties of Red Blood Cell Deformability. Front. Physiol. 2018, 9, 656. DOI:10.3389/fphys.2018.00656 [Google Scholar]
-
Danielczok JG, Terriac E, Hertz L, Petkova-Kirova P, Lautenschläger F, Laschke MW, et al. Red Blood Cell Passage of Small Capillaries Is Associated with Transient Camediated Adaptations. Front. Physiol. 2017, 8, 979. DOI:10.3389/fphys.2017.00979 [Google Scholar]
-
Cahalan SM, Lukacs V, Ranade SS, Chien S, Bandell M, Patapoutian A. Piezo1 links mechanical forces to red blood cell volume. Elife 2015, 4, e07370. DOI:10.7554/eLife.07370 [Google Scholar]
-
Retailleau K, Duprat F, Arhatte M, Ranade SS, Peyronnet R, Martins JR, et al. Piezo1 in Smooth Muscle Cells Is Involved in Hypertension-Dependent Arterial Remodeling. Cell Rep. 2015, 13, 1161–1171. DOI:10.1016/j.celrep.2015.09.072 [Google Scholar]
-
Choi D, Park E, Jung E, Cha B, Lee S, Yu J, et al. Piezo1 incorporates mechanical force signals into the genetic program that governs lymphatic valve development and maintenance. JCI Insight 2019, 4, e125068. DOI:10.1172/jci.insight.125068 [Google Scholar]
-
Matic Z, Catrambone V, Valenza G. Neural mechanisms underlying blood pressure dynamics and cardiovascular control. Am. J. Physiol. Heart Circ. Physiol. 2026, 330, H1553–H1577. DOI:10.1152/ajpheart.00803.2025 [Google Scholar]
-
Zheng M, Yao Y, Borkar NA, Thompson MA, Zhang EY, Vogel ER, et al. Piezo Channels Modulate Stretch-induced Remodeling and Calcium Regulation in Human Lung Fibroblasts. Am. J. Resp. Crit. Care 2024, 209. DOI:10.1164/ajrccm-conference.2024.209.1_MeetingAbstracts.A7191 [Google Scholar]
-
Valenza G, Matic Z, Catrambone V. The brain-heart axis: integrative cooperation of neural, mechanical and biochemical pathways. Nat. Rev. Cardiol. 2025, 22, 537–550. DOI:10.1038/s41569-025-01140-3 [Google Scholar]
-
Pirri C. PIEZO Channels in Mechano-Inflammation: Gatekeepers of Neuroimmune Crosstalk. Diseases 2025, 13, 263. DOI:10.3390/diseases13080263 [Google Scholar]
-
Darkow E, Yusuf D, Rajamani S, Backofen R, Kohl P, Ravens U, et al. Meta-Analysis of Mechano-Sensitive Ion Channels in Human Hearts: Chamber- and Disease-Preferential mRNA Expression. Int. J. Mol. Sci. 2023, 24, 10961. DOI:10.3390/ijms241310961 [Google Scholar]
-
Picard V, Guitton C, Thuret I, Rose C, Bendelac L, Ghazal K, et al. Clinical and biological features in PIEZO1-hereditary xerocytosis and Gardos channelopathy: A retrospective series of 126 patients. Haematologica 2019, 104, 1554–1564. DOI:10.3324/haematol.2018.205328 [Google Scholar]
-
Ranade SS, Syeda R, Patapoutian A. Mechanically Activated Ion Channels. Neuron 2015, 87, 1162–1179. DOI:10.1016/j.neuron.2015.08.032 [Google Scholar]