1. Introduction
The progression of fast-developing electronic devices, green vehicles and clean energy sources has emerged demand for next-generation energy storage devices with efficiency and stability. Conventional Li-ion batteries with liquid electrolytes have been widely used in various energy storage sectors; however, they face risks of thermal instability and have a low safety profile. Thus, efforts have been made to enhance safety and power density. These include replacing liquid electrolytes with solid-state alternatives. Researchers are also working to increase operating voltage by optimizing the surface area of active materials. Another approach involves modifying the formulation of existing conductive additives. Moreover, efforts have been extended towards developing high-energy density rechargeable electric batteries. Major advancement can be achieved by creating an optimal combination of electrodes, solid electrolytes and efficient ways of utilization with extreme thermal and mechanical stability for multiple charge-discharge cycles. Development of an ideal battery concept requires various key ingredients that facilitate energy transmission at a rate applicable in specific devices (microscale), together with overall low thermal and mechanical instability. Researchers have also reported a thin-film battery suitable for mass-electricity storage. This battery possesses an active area energy density of nearly 0.5 Wh. Its energy density is approximately 120 times greater than a comparable solid electrolyte configuration. However, Research on light-weight structure designs and practical commercial applications has revealed several promising trends in energy storage technology. Many of these applications show strong scalability potential, particularly for handling extremely high currents. They also demonstrate low total power consumption while maintaining high stability. Among the most significant advances are innovative low-resistance flow batteries for renewable energy storage. Another key development involves all-solid-state batteries featuring novel surface liquid electrolytes that enable higher current transport. These batteries offer large discharge power and high electrode activity. Self-healing alloy batteries also represent a major breakthrough in current energy storage technology. Meanwhile, researchers are making progress in developing multi-scale magnetorheological elastomer composites, which are finding increasingly extensive applications in energy storage systems. However, flammable ingredients, electrolyte infiltration, and thermal runaway limit their suitability for next-generation devices. Solid-state batteries have received more interest as a promising prospect as an alternative thanks to the inherent safety structure, broad operating temperature and their superior electrochemical stability. Due to its role as the critical design factor in a successful battery, the solid-state electrolyte must optimize a host of competing factors: high ion conductivity, mechanical robustness, chemical stability with respect to the active materials in operation, good electrode material interfaces, and so on.
Sulfide solid state electrolytes (SSEs) are considered a very promising candidate with high ionic conductivity, between 10−3–10−2 S cm−1, compared to many other SSEs. Although the advantage of sulfide SSEs over others, their electrochemical stability windows are limited, reactions with humid gas, unstable interface with electrodes, etc. In order to provide a complementary part of this, exhaustive efforts are carried out by doing the theoretical modelling and simulations of SSEs composed of sulfides for unravelling their structural, as well as functional properties, to understand the fundamental properties in facilitating the experimental development and optimization of the materials.
Quantum mechanical based advanced computational techniques, for example, ab initio calculations and molecular dynamics simulations, are exceptionally useful methods to explore the atomic-level structure-function relations of solid-state electrolytes. Such machine-learning based predictions allow for quantitative measurements of salient properties such as mechanical strength, ion transport behavior, and thermodynamic stability, and thus pave the route toward linking theory with experiments. These calculations can also be used as predictors in the exploration and development of next-generation electrolytes based on systematic material screening.
Quantum chemistry is combined with computational thermodynamics methods to get an overall evaluation of the system stability, making it possible to establish phase diagrams and to draw the convex hull. The determination of the electrochemical window using first-principles calculations consists of several analyses—stoichiometric decomposition analysis, electronic structure analysis utilizing orbital energy gap and phase stability as a function of the electric potential. These multi-scale modeling approaches establish valuable design guidelines. They enable the development of high-performance solid-state electrolytes (SSEs) with precisely tailored properties. This methodology significantly accelerates the materials development cycle for electrochemical energy storage (EES) systems. The advancement is achieved through the systematic implementation of material-by-design strategies.
Ultimately, computational modeling and simulating of sulfide-based SSEs could greatly facilitate the development of solid-state batteries, as shown in this review, improving our understanding of fundamental properties as well as accelerating the discovery and optimization of new materials. This highlights the importance of theory calculations for materials engineering, and specifically for sulfide electrolytes. Theory calculation provides understanding of the correlation between the material structure and properties, makes predictions for experimental results, and guides experimental efforts. The above efforts give a guide for further engineering high-performance solid-state batteries.
2. Sulfide Electrolytes and Theoretical Calculations
2.1. Research History of Sulfide Electrolytes
The investigation into sulfide electrolytes dates back to the 1960s with the utilization of β-alumina in high-temperature sodium-sulfur batteries. This event signaled the commencement of research into solid-state ionic conductors [
1]. Following the initial development of oxide solid electrolytes, their application in all-solid-state lithium-ion batteries (ASSLBs) was constrained due to their inherently low ionic conductivity. This limitation prompted further exploration into alternative materials. Subsequently, the research groups led by Tatsumisago and Kanno made significant strides by investigating a range of sulfide-based electrolytes. Their findings not only marked a pinnacle in the quest for high-performance ion conductors but also sparked a surge of interest in the advancement of ASSLBs [
2,
3,
4]. These sulfide-based electrolytes exhibit an impressive level of ionic conductivity, rivaling that of conventional organic liquid electrolytes. This characteristic positions them as top contenders for use in ASSLBs. In recent years, the research on sulfide electrolytes has made significant progress. For example, the sulfide solid electrolyte developed by Kanno’s team has an ionic conductivity (2.5 × 10
−2 S cm
-1) higher than that of liquid electrolytes [
2,
3]. The findings from these studies have broadened the potential for ASSLBs to be utilized in applications like electric vehicles and portable electronics. However, there are still hurdles to overcome, including limitations in the electrochemical stability range, inadequate chemical interaction with electrode materials, and suboptimal mechanical characteristics.
2.2. Current Research Progress
Currently, the studies on sulfide electrolytes focus on enhancing their ionic conductivity, tuning the structure to maximize the interfacial stability with an electrode, and searching for novel synthesis routes. For instance, sulfide electrolytes are synthesized by mechanical ball milling, high-temperature solid phase reaction, and liquid phase synthesis to achieve higher ionic conduction and better electrode/electrolyte interface contact [
5,
6,
7]. Meanwhile, Researchers are enhancing sulfide electrolytes through element doping and structure regulation. These modifications improve the electrolytes’ electrochemical and chemical stability. Such improvements make the electrolytes more suitable for real-world application conditions. The optimized sulfide electrolytes demonstrate superior electrochemical performance. Consequently, their application in all-solid-state lithium-sulfur batteries has gained significant attention. Current research focuses on three key areas: increasing sulfur cathode utilization rates, suppressing the polysulfide shuttle effect, and optimizing overall battery structure and performance.
2.3. Different Types of Sulfide Electrolytes
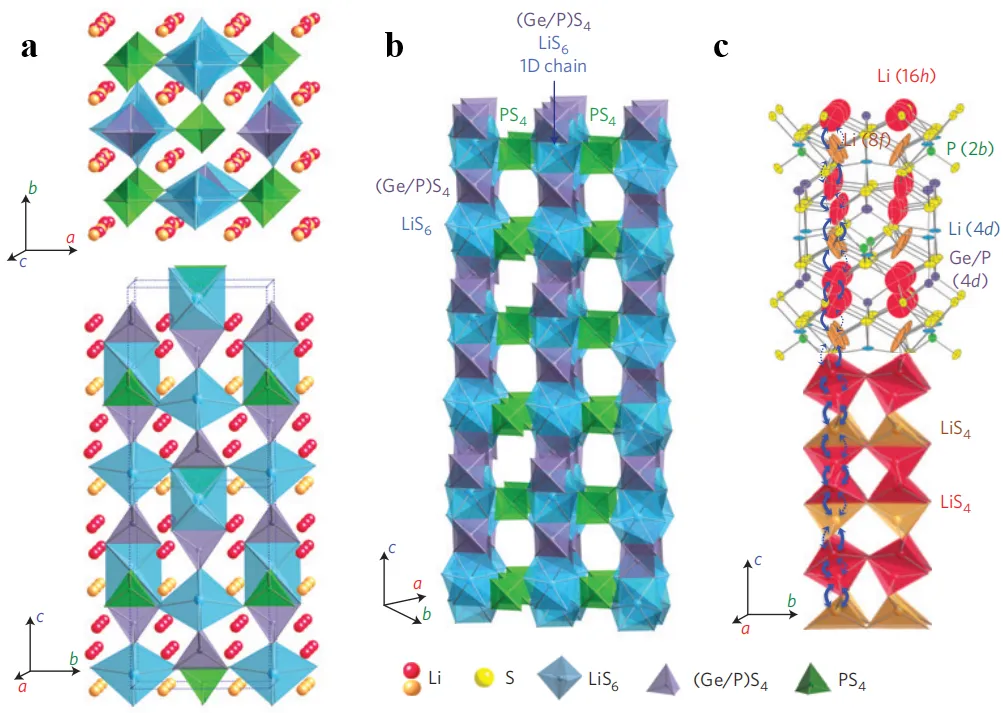
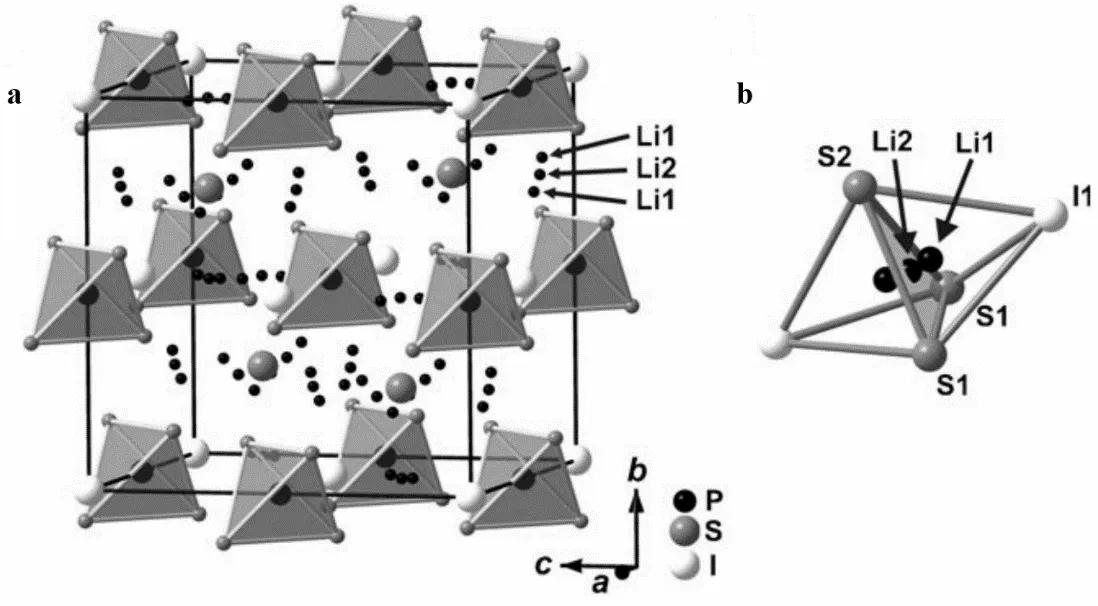
Sulfide electrolytes can be divided into two categories according to their composition: binary and ternary. Binary sulfide electrolytes are mainly composed of Li
2S and P
2S
5, such as Li
3PS
4 and Li
7P
3S
11. This type of electrolyte can achieve higher ionic conductivity by controlling the ratio of Li
2S and P
2S
5 and the heat treatment temperature. For example, after appropriate heat treatment, the ionic conductivity of 80Li
2S–20P
2S
5 and 75Li
2 S–25P
2S
5 glass-ceramic materials is improved to 7.2 × 10
−4 S cm
−1 and 2.8 × 10
−4 S cm
−1, respectively [
6]. Ternary sulfide electrolytes introduce other components such as MS
2 (M = Si, Ge, Sn) on the basis of binary electrolytes, such as Li
10GeP
2S
12(LGPS) [
2] and Li
6PS
5X (X = Cl, Br, I) [
7] and so on. Ternary sulfide electrolytes further improve ionic conductivity and electrochemical stability through element substitution and structural regulation, as shown in
. Taking Li
10GeP
2S
12 as an example, it has a unique three-dimensional framework structure composed of (Ge
0.5P
0.5)S
4 tetrahedron, PS
4 tetrahedron, LiS
4 tetrahedron and LiS
6 octahedron, showing extremely high ionic conductivity (1.2 × 10
−2 S·cm
−1). The 1D lithium-ion conduction pathway arises from edge-sharing LiS₄ tetrahedra at 16h and 8f sites, forming tetrahedral chains connected via corner-sharing (
c). Neutron diffraction reveals lithium at these sites exhibits highly anisotropic thermal vibrations, displacing toward interstitial positions between 16h-16h and 16h-8f sites. This displacement directly evidences 1D ion migration along the c-axis. Partial occupancy (0.691(5) at 16h; 0.643(5) at 8f) reflects lithium’s average distribution along the pathway—a hallmark of superionic conductors. In addition to the above two categories, there are some special sulfide electrolyte structure types, such as thio-LISICON type, tetragonal LGPS type and argyrodite Li
6PS
5X type. Thio-LISICON electrolytes are derived by replacing oxygen in LISICONγ-Li
3PO
4 solid electrolytes with sulfur, like (100−x)Li
2S–xP
2S
5, (100−x)Li
2S–xSiS
2, and Li
4−xGe
1−xP
xS
4 (0 < x < 1) [
2]. Since the electronegativity of sulfur is lower than that of oxygen, this type of electrolyte reduces the binding energy of lithium ions. It increases the ion migration channel, thereby promoting the migration of lithium ions and having high ionic conductivity. Tetragonal LGPS electrolytes are represented by Li
10GeP
2S
12, whose structure contains (Ge
0.5P
0.5)S
4 tetrahedrons and LiS
6 octahedrons connected by sharing edges to form one-dimensional chains. These one-dimensional chains are then connected to each other by PS
4 tetrahedrons to form a three-dimensional framework, providing abundant migration channels for lithium ions. Taking Li
6PS
5I as an example of [
7], its structure is composed of 136 tetrahedrons formed by S atoms, P atoms fill four of the tetrahedral holes, I ions are located in the corners and faces of the unit cell, and lithium ions occupy part of the tetrahedral holes formed by S
2− and I
− in a disordered pattern, showing relatively low ionic conductivity, as shown in
. But, its ionic conductivity can be significantly improved through structural optimization and element substitution, like Li
6 PS
5Cl.
. (<strong>a</strong>) Architecture and Lithium Ion Conduction: Schematic illustration of the architecture and lithium ions participating in the conduction; (<strong>b</strong>) Architecture: Schematic representation of the Li<sub>10</sub>GeP<sub>2</sub>S<sub>12</sub> architecture, the one dimensional (1D) chains of LiS<sub>6</sub> octahedra and (Ge<sub>0.5</sub>P<sub>0.5</sub>)S<sub>4</sub> tetrahedra connected via the shared edges. These chains are connected by the corners of PS<sub>4</sub> tetrahedra; (<strong>c</strong>) Li Ion Conduction Pathway: The zigzag path for the lithium ion hop along the <em>c</em>-axis is visualized. This conduction is governed by the lithium ions residing at LiS<sub>4</sub> tetrahedra at 16h and 8f sites. The thermal ellipsoid is taken at 30% probability. The anisotropic thermal vibration of the lithium ions at those tetrahedral sites makes room for the one-dimensional conduction pathway. Reproduced with permission of Ref. [<a href="#B2" class="html-bibr">2</a>], Copyright 2011 Nature Materials.
. (<strong>a</strong>) Crystal Structure Section: A segment of the Li<sub>6</sub>PS<sub>5</sub>I crystal structure illustrating the arrangement of PS<sub>4</sub> tetrahedra, along with the distribution of S<sub>2</sub> ions (located in four octants of the unit cell) and I ions (positioned at the corners and faces of the unit cell); (<strong>b</strong>) Face-Sharing Tetrahedron: A depiction of the S<sub>3</sub>I<sub>2</sub> double tetrahedron, where the shared face contains Li1 ions (positioned above and below the common face) and Li2 ions (located at the center of the shared face). Reproduced with permission of Ref. [<a href="#B7" class="html-bibr">7</a>], Copyright 2008 Angewandte Chemie.
2.4.1. First Principles Calculation
First-principles calculation is an atomic-scale simulation method based on the basic principles of quantum mechanics. It predicts material properties from the electronic level by solving the Schrödinger equation and can describe the interaction between atoms without relying on empirical parameters. Among them, density functional theory (DFT) is the most commonly used calculation framework. By mapping the ground state properties of a multi-body quantum system into an electron density functional and using the approximate solution of the Kohn-Sham equation, it can accurately predict key performance parameters such as the electronic structure and energy distribution of the material. In the study of SSEs, DFT has been widely used in the theoretical prediction of mechanical properties, including the calculation of key parameters such as shear modulus (G), bulk modulus (B), Poisson’s ratio (ν) and Young’s modulus (E) [
8]. These parameters have important guiding values for evaluating the mechanical stability of SSEs and the safety of battery operation.
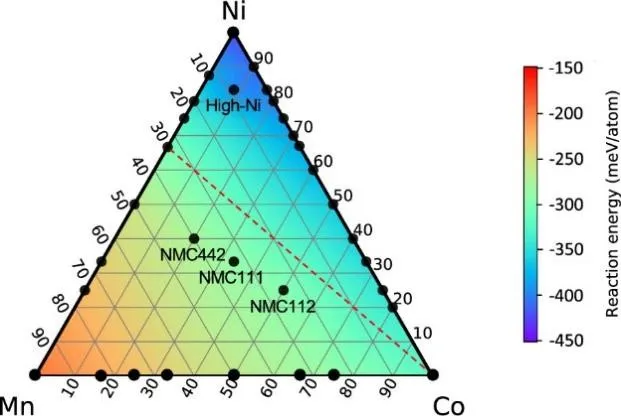
Studies have shown that the shear modulus of oxide-based SSEs obtained through DFT calculation is generally higher than that of traditional liquid electrolytes. This property gives them excellent resistance to shear deformation and helps to inhibit lithium dendrite penetration. It is worth noting that DFT has shown unique advantages in the study of interface stability. Hideyuki Komatsu’s team [
9] used DFT calculations to systematically study the interface reaction mechanism between layered LiNi
xMn
yCo
1−x−yO
2 (NMC) positive electrode materials and sulfide electrolyte Li
6PS
5Cl (LPSCl). The calculation results show that when the nickel content x value increases from 0.33 to 0.80, the interface reaction energy barrier decreases by about 0.4 eV, significantly increasing the activity of the interface side reaction. The reduction in Co content may increase the risk of side reactions. It is worth noting that the study quantified the interface stability trend (Mn > Co > Ni) by constructing a ternary phase diagram (Ni-Mn-Co) and pointed out that Li
2CO
3, NiO surface phase and LiNbO
3 buffer layer can be used as an effective interface passivation layer due to their limited reactivity with NMC/LPSCl (ΔE
mutual > −100 meV atom
−1), as shown in
. This work also used a pseudo-binary phase diagram to analyze the interface volume change (shrinkage rate > 30%), revealing the contact failure mechanism of high-nickel positive electrode under high pressure, and providing a theoretical basis for composition optimization. This discovery reveals the regulatory mechanism of transition metal element ratio on interface stability, and provides a theoretical basis for interface engineering of high-nickel positive electrodes. At the same time, the study also found that solid electrolyte interphase (SEI), such as Li
2CO
3 and LiNbO
3 buffer layer, shows low reactivity with the positive electrode/electrolyte system, which points out the direction of material selection for designing new interface protection layers.
. Ternary contour plot of reaction energies at the LMO/LPSCl interface. The heat map is obtained by interpolating the calculated reaction energies of the labeled components. The red dashed line connecting LCO and LiNi<sub>0.7</sub>Mn<sub>0.3</sub>O<sub>2</sub> shows the trend line for the cathode active material with lower Co to maintain the same chemical stability. Reproduced with permission of Ref. [<a href="#B9" class="html-bibr">9</a>], Copyright 2022 American Chemical Society.
2.4.2. Molecular Dynamics Simulation (MD)
Molecular dynamics (MD) simulation is based on Newton’s laws of motion. It numerically solves the equations of motion of atoms or molecules, tracks the evolution of the system over time, and then obtains thermodynamic properties (such as free energy, diffusion coefficient) and kinetic information (such as ion migration path). According to the choice of potential function, MD methods can be divided into two categories: (1) classical MD, which approximates the interaction between atoms through empirical potential functions (such as Lennard-Jones potential and Morse potential), has high computational efficiency, and is suitable for large systems and long-term simulations; (2) ab initio molecular dynamics (AIMD), which calculates the forces on atoms based on quantum mechanics, has higher accuracy but is computationally expensive, and is often used for electronic structure coupling analysis of small systems and short time scales [
10].
In the study of grain boundary effects in solid electrolytes, classical MD has become a mainstream tool due to its high efficiency. Yu et al. [
11] systematically simulated the structural characteristics of three low-energy symmetrical tilted grain boundaries (Σ5(310), Σ5(210) and Σ3(112)) and their effects on Li⁺ diffusion in the grain boundary ion transport mechanism of lithium lanthanum zirconium oxide (Li
7La
3Zr
2O
12, LLZO). By constructing a model system containing 160 to 96 LLZO unit cells and using a force field based on bond valence parameters to describe atomic interactions, the study revealed that the Li⁺ diffusion coefficient in the grain boundary region is significantly lower than that in the bulk phase. The inhibition effect is closely related to the grain boundary structure: the dense Σ3 grain boundary only reduces the diffusion rate by about 50% because the activation energy is comparable to that of the bulk phase (0.52 eV); while the activation energy of the loose Σ5 grain boundary increases by 35% (to 0.71 eV), resulting in a sudden drop of 2 orders of magnitude in the diffusion rate at room temperature. This discovery provides an atomic-scale explanation for the differences in grain boundary resistance in experimental observations (e.g., grain boundary resistance in high-temperature sintered samples accounts for only 8%). In addition, by analyzing the lattice stress distribution and atomic trajectories, the researchers further discovered that there is diffusion anisotropy within the Σ3 grain boundary—the diffusion rate along the crystal interface even exceeds that of the bulk phase, while cross-interface diffusion is severely hindered. This type of work highlights the unique value of MD simulation in correlating microstructure with macroscopic transport properties.
2.4.3. Ab Initio Molecular Dynamics (AIMD)
AIMD combines quantum mechanical calculations with molecular dynamics to obtain the potential energy of interactions between atoms by solving the Kohn-Sham equation in real time without relying on empirical potential functions. This first-principles method can accurately describe the evolution of electronic structure and the dynamic reconstruction of chemical bonds, and is particularly suitable for complex systems involving charge transfer, reaction path exploration,
etc. (such as electrochemical interfaces, phase change processes). Compared with classical MD, AIMD has significant advantages in chemical accuracy and prediction of unknown system properties, but its high computational cost limits the scale of simulation systems (usually <1000 atoms) and time scale (usually <100 ps).
In the study of ion transport mechanisms in SSEs, AIMD has become a key tool due to its high precision. Mo et al. [
12] used AIMD simulation to simulate the Li⁺ diffusion behavior in the superionic conductor Li
10GeP
2S
12 (LGPS). They revealed the cooperative migration mechanism: multiple Li
+ migrate synchronously in a non-localized manner through dynamic channels, rather than the traditional isolated ion hopping model. The study calculated that the activation energy of Li
+ is 0.21 eV, which is highly consistent with the experimental value (0.22 eV), and predicted that the room temperature ionic conductivity is 14.2 mS cm
−1(experimental value: 12 mS cm
−1). This work not only verifies the reliability of AIMD in quantifying transport parameters but also guides the design of new SSEs (such as doping to optimize channel connectivity).
3. Interface Issues between Sulfide Electrolytes and Electrode Materials
3.1. Interface Stability Issues
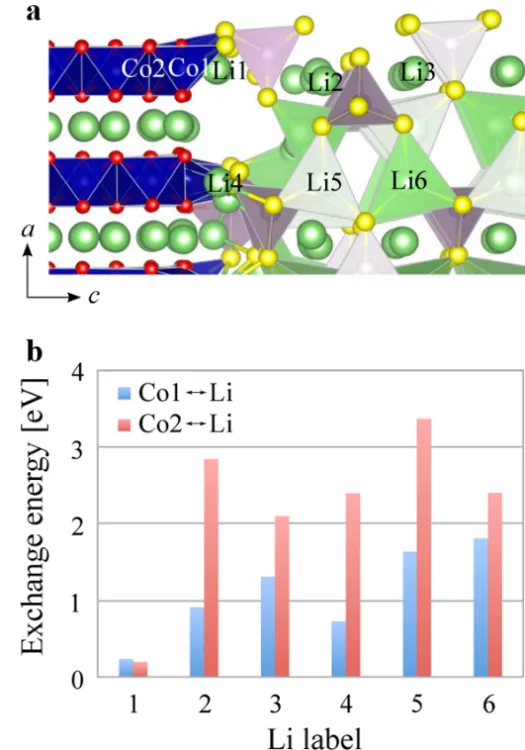
When sulfide solid electrolyte comes into contact with electrode materials, chemical reactions easily occur to form an interface layer, which leads to electrolyte instability. Haruyama et al. [
13] studied the cation diffusion, they discussed the interface characteristics of the LiCoO
2/β−Li
3PS
4 in detail by the density functional theory (DFT) calculations. They found that the exchange energy of elements of Co and P are negative, and this means that it is easy for the elements to diffuse at the interface and a thermodynamically stable interface layer will be formed because of the element diffusion, while the ionic conductivity of the interface layer is low and the interface impedance increases significantly. In particular, Co and P exchange energy values are −1.2 eV and −0.8 eV, respectively, suggesting that the diffusion process of those elements at the interface possesses a strong driving force thermodynamically, as shown in
.
. (<strong>a</strong>) Interface Composition: The interface of LCO (110)-LPS (010). Different colours indicate Li (light green), O (red), P (purple), S (yellow), and Co (blue). Polyhedra in the Figure represent the CoO<sub>6</sub> (blue), PS<sub>4</sub> (purple) and LiS<sub>4</sub> (lightgreen) complexes. (<strong>b</strong>) Exchange Energy: The plot of exchange energy vs. the Li label, where the <em>x</em>-axis is the label of Li and the <em>y</em>-axis is in eV. Reproduced with permission of Ref. [<a href="#B13" class="html-bibr">13</a>], Copyright 2016 American Chemical Society.
In addition, some sulfide electrolytes show thermodynamic and kinetic instability towards metallic lithium. When Li
10GeP
2S
12 [
14] is used to generate interface products such as Li
2S and Li
3P, as shown in
. The formation of these products not only increases the interface resistance but also affects the overall performance of the battery. Experimental data show that after Li
10GeP
2S
12 comes into contact with lithium metal, the interface resistance increases from the initial 10 Ω cm
2 to about 50 Ω cm
2, significantly reducing the charge and discharge efficiency of the battery.
. Schematic diagram of the interaction between lithium metal and sulfides, phosphides, and arsenides in SSEs. Reproduced with permission of Ref. [<a href="#B14" class="html-bibr">14</a>], Copyright 2018 Elsevier.
When the oxide positive electrode contacts the sulfide solid electrolyte, due to the significant difference in electrochemical potential between the two, lithium ions tend to migrate from the sulfide electrolyte to the oxide positive electrode, forming a space charge layer (SCL). The ion concentration gradient in this area not only significantly increases the interface resistance but also inhibits the kinetic performance of lithium ion migration. Takada et al. [
15] modified the interface between LiCoO
2 and Li
3.25Ge
0.25P
0.75S
4 electrolyte by LiNbO
3 coating, and found that the interface resistance of the uncoated electrode was as high as 1 × 10
4 Ω, while the resistance after coating was reduced by two orders of magnitude, indicating that the space charge layer (thickness of only a few nanometers) plays a decisive role in the interface impedance, as shown in
. It is worth noting that although the physical thickness of the space charge layer is limited, the local lithium-ion depletion effect caused by it will significantly hinder carrier transport. In addition, the volume change of the positive electrode material during the charge and discharge process, such as the lattice shrinkage of LiNi
0.8Co
0.1Mn
0.1O
2 when de-lithiation occurs, will lead to mechanical contact loss between the particles and the electrolyte, further deteriorating the interface impedance. Koerver et al. [
16] compared the morphology of the interface between NCM-811 and β-Li
3PS
4 before and after charge and discharge and found that micron-scale cracks appeared at the interface after cycling, confirming the direct correlation between the volume effect and interface degradation, as shown in
b,c.
. (<strong>a</strong>) Complex impedance diagram of In-Li//LiMn<sub>2</sub>O<sub>4</sub> battery. Reproduced with permission of Ref. [<a href="#B15" class="html-bibr">15</a>], Copyright 2012 Elsevier. (<strong>b</strong>,<strong>c</strong>) SEMs of a Li−In|β-Li<sub>3</sub>PS<sub>4</sub>|NCM-811/β-Li3PS4 cell after single charging to 4.3 V <em>vs</em>. Li/Li<sup>+</sup> at 0.1 C. Reproduced with permission of Ref. [<a href="#B16" class="html-bibr">16</a>], Copyright 2017 American Chemical Society.
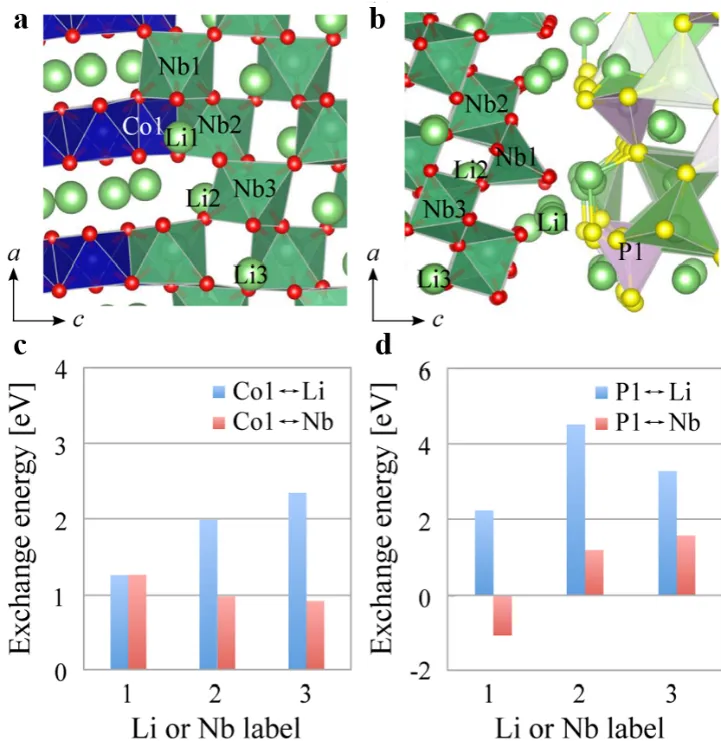
Theoretical calculation simulation provides key insights into the interface reaction mechanism and optimization design of solid-state batteries. Taking the oxide cathode/sulfide solid electrolyte interface as an example, Haruyama et al. [
13] systematically studied the cation diffusion behavior at the interface of LiCoO
2 (LCO) and β−Li
3PS
4 (LPS) by first-principles calculation. Their results showed that exchange energy between Co and P (ΔE < 0) is strongly negative (about −2.18 eV), which means that the Co and P at the interface is a thermodynamically spontaneous process. The formation of interface states can be attributed to the stabilization of their electronic structure, as shown in
. As a result, this interdiffusion behavior not only decreases the formation energy of lithium vacancy in the interface and aggravates the growth of the lithium depletion layer, but also causes the high interface impedance and cycle performance degradation problem in experiments. It should be mentioned that the research further showed that the addition of a LiNbO
3 (LNO) buffer layer can effectively inhibit Co diffusion, since the exchange energy between Co and Nb is substantially increased to +1 eV, which offers important theoretical guidance for interface engineering, as shown in
c.
. (<strong>a</strong>) Interface Structures: Depiction of the LCO (110)/LNO (110) and LNO (110)/LPS (010) interfaces. Nb atoms are shown as green spheres, and NbO6 complexes are represented by green polyhedra; (<strong>b</strong>) Exchange Energies: Analysis of the exchange energies at the LCO/LNO interface; (<strong>c</strong>) Co↔Li/Nb exchange, and at the LNO/LPS interface; (<strong>d</strong>) P↔Li/Nb exchange. Reproduced with permission of Ref. [<a href="#B13" class="html-bibr">13</a>], Copyright 2016 American Chemical Society.
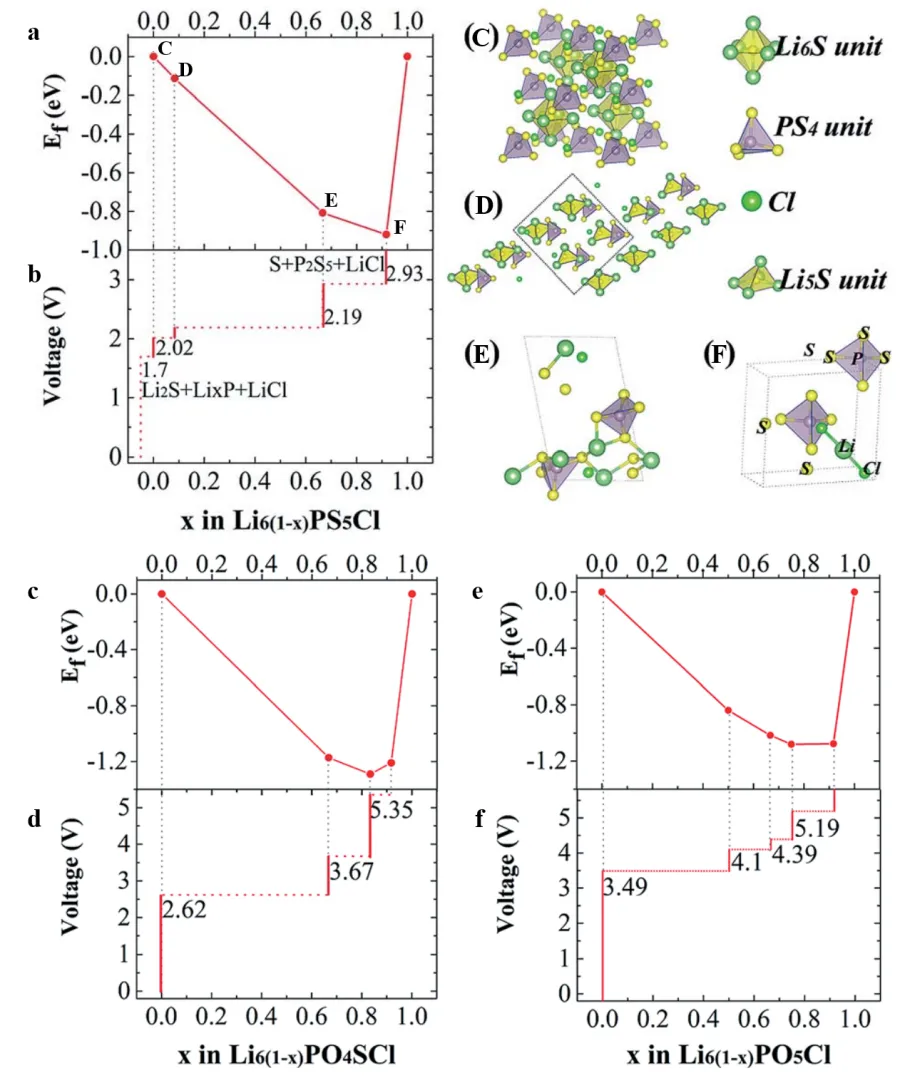
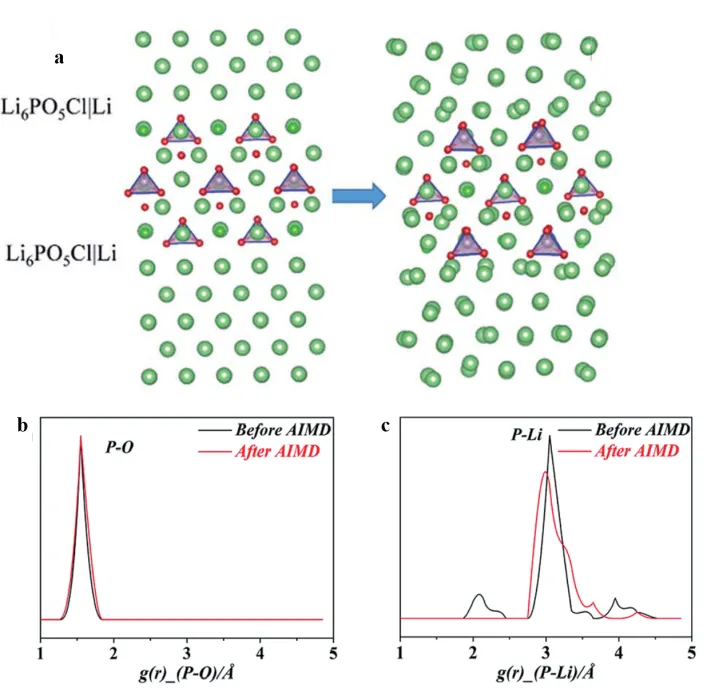
Theoretical calculations also show an important guiding value in optimizing the interfacial stability of sulfide solid electrolytes. Xu et al. [
17] systematically studied the electrochemical stability of the argyrodite-type Li
6PA
5Cl (A = O/S) electrolyte system and revealed the key role of oxygen doping in inhibiting interfacial reactions. The electrochemical potential diagrams of Li
6PO
5Cl, Li
6PO
4SCl and Li
6PS
5Cl constructed by them are shown in
, which shows that the electrochemical windows of the three are 0–3.49 V, 0–2.62 V and 1.7–2.19 V (
vs. Li/Li
+), respectively. Among them, Li
6PO
5Cl shows the widest electrochemical window due to the stabilization effect of the strong P-bond. Its oxidation potential is about 1.3 V higher than that of Li
6PS
5Cl, which significantly broadens its compatibility with high-voltage positive electrodes (such as Li
0.25MnO
2, with an operating voltage of 2.72–3.65 V). AIMD simulations further verified the structural stability of the Li
6PO
5Cl|Li interface at 600 K, as shown in
a. The radial distribution functions of P-O and P-Li, showed no significant changes, indicating that oxygen doping can effectively inhibit the reduction and decomposition of sulfide electrolytes and the formation of low-conductivity phases such as Li
3P/Li
2S, as shown in
b,c.
. (<strong>a</strong>) Convex hull for Li<sub>6(1-x)</sub>PS<sub>5</sub>Cl (0 < x < 1); (<strong>b</strong>) Electrochemical potentials of Li<sub>6(1−x)</sub>PS<sub>5</sub>Cl structures. Inset structures: (C) Li<sub>6</sub>PS<sub>5</sub>Cl, (D) Li<sub>5.5</sub>PS<sub>5</sub>Cl, (E) Li<sub>2</sub>PS<sub>5</sub>Cl, (F) Li<sub>0.5</sub>PS<sub>5</sub>Cl. The electrochemical window of Li<sub>6</sub>PS<sub>5</sub>Cl is only 0.49 V. (<strong>c</strong>) Convex hull and (<strong>d</strong>) electrochemical potentials for Li6(1−x)PO4SCl. (<strong>e</strong>) Convex hull and (<strong>f</strong>) electrochemical potentials for Li<sub>6(1−x)</sub>PO<sub>5</sub>Cl. Substituting S with O increases the electrochemical window from 2.62 V to 3.49 V. Reproduced with permission of Ref. [<a href="#B17" class="html-bibr">17</a>], Copyright 2019 The Royal Society of Chemistry.
. (<strong>a</strong>) Slab model for characterizing the interface of Li<sub>6</sub>PO<sub>5</sub>Cl|Li; Pairwise correlation functions (PCFs) before (black line)/after (red line) AIMD simulation (90 ps at 600 K): (<strong>b</strong>) P-O and (<strong>c</strong>) P-Lipcf for Li<sub>6</sub>PO<sub>5</sub>Cl|Li interface. Reproduced with permission of Ref. [<a href="#B17" class="html-bibr">17</a>], Copyright 2019 The Royal Society of Chemistry.
Understanding the dynamic evolution of interfacial reactions in sulfide-based solid-state batteries requires the integration of advanced in situ characterization techniques and theoretical simulations. Yamagishi et al. [
18] achieved the first real-time visualization of lithium distribution and interface degradation in NCA/LPS composite electrodes using operando time-of-flight secondary ion mass spectrometry (TOF-SIMS). By mapping Li-containing fragments (e.g., 6Li
+, Li
2O
+) and degradation products (PO
x−, SO
x−), this technique revealed that during charging, the 6Li
+ signal intensity decreased in NCA particles, but some particles retained high Li concentration due to insufficient electron/ion pathways. This indicates that the microstructure of composite electrodes needs optimization to reduce “inactive particles”.
Under high-voltage stress (5–6 V), the Po
x− fragment intensity monotonically increased, corresponding to the oxidation of LPS to form insulating phases such as Li
3PO
4. In contrast, the SO
3− fragment exhibited reversible redox behavior, suggesting dynamic polymerization/depolymerization reactions of sulfides at the interface. Additionally, Auvergniot et al. confirmed the redox activity of Li
3PS
4Cl electrolytes during cycling using XPS [
19]. Sakuda et al. directly observed crack propagation and phase transitions at the LiCoO
2/Li
3PS
4 interface through in situ TEM [
20], while Xu et al. employed AIMD simulations to validate the mechanism of oxygen doping in suppressing decomposition at the Li
6PO
5Cl|Li interface [
21]. These findings collectively highlight the importance of complementary techniques in elucidating interfacial mechanisms.
4. Ion Migration and Diffusion Behavior
Ion migration and diffusion behaviors are important factors to study SSEs and deeply influence the performance of batteries, including charge and discharge rate, cycle life and energy density. These important behaviors are significant for high-performance solid-state batteries’ design.
4.1. ML Potential Energy Surface
Machine learning (ML) potential energy surfaces are constructed based on machine learning methods and are used to describe the potential energy surface of atomic interactions. Common construction methods include Gaussian approximate potentials (GAPs) [
22], neural network potentials (NNPs) [
23,
24,
25,
26],
etc. These methods can predict the energy and force of atomic interactions with high accuracy by learning from a large amount of first-principles calculation data. Compared with traditional empirical potentials, ML potential energy surfaces have higher accuracy and better transferability, and can more accurately describe complex chemical environments and multi-body interactions. GAP Gaussian process regression-based machine learning technique that constructs potential energy surfaces by fitting the relation between interatomic distances and energies. The benefit of GAP is that it can deal with complex potential energy surfaces and could be very accurate in the prediction of new structures. For instance, when studying the sulfide electrolytes, GAP could be used to simulate the migration route and energy barrier of ions in the lattice. NNP uses the excellent fitting function of neural networks to build a potential energy surface. By training a lot of atomic configurations and their corresponding energy data, NNP could learn the complex potential energy relationship and have excellent generalization ability when predicting a new configuration. In sulfide electrolytes research, we can adopt NNP to do the structural optimization, predict ionic conductivity,
etc.
4.2. Application of MLMD in Sulfide Electrolyte Research
Machine learning molecular dynamics (MLMD) combines machine learning potential energy surfaces and molecular dynamics simulations to study ion migration and diffusion behavior on a large scale and over a long time scale. Through MLMD simulation, the diffusion coefficient and activation energy of ions in sulfide electrolytes can be calculated [
23]. Fu et al.[
27] used MLMD to study the diffusion behavior of Li ions in Li
10GeP
2S
12 and found that its activation energy was consistent with the experimental value, indicating that MLMD can accurately predict ionic conductivity. In addition, Pei et al. [
28] and Li et al. [
29] used MLMD to study the ion diffusion behavior in antiperovskite Li
3OX (X = Cl or Br) and Na
3OBr, respectively, and found that the diffusion coefficient was linearly related to the defect concentration. MLMD can also be used to optimize the material structure of sulfide electrolytes. Wang et al. [
30] and Xu et al. [
31] used MLMD to study the structure and phase transition behavior of Li
3YCl
6 and Li
3ErCl
6, respectively, and found that these materials undergo phase transitions at around 460 K, resulting in a significant decrease in activation energy and exhibiting nonlinear Arrhenius behavior. These research results provide an important theoretical basis for optimizing material structure.
5. Ion Conduction Mechanism of Sulfide Electrolytes
5.1. Ionic Conductivity of Sulfide Electrolytes
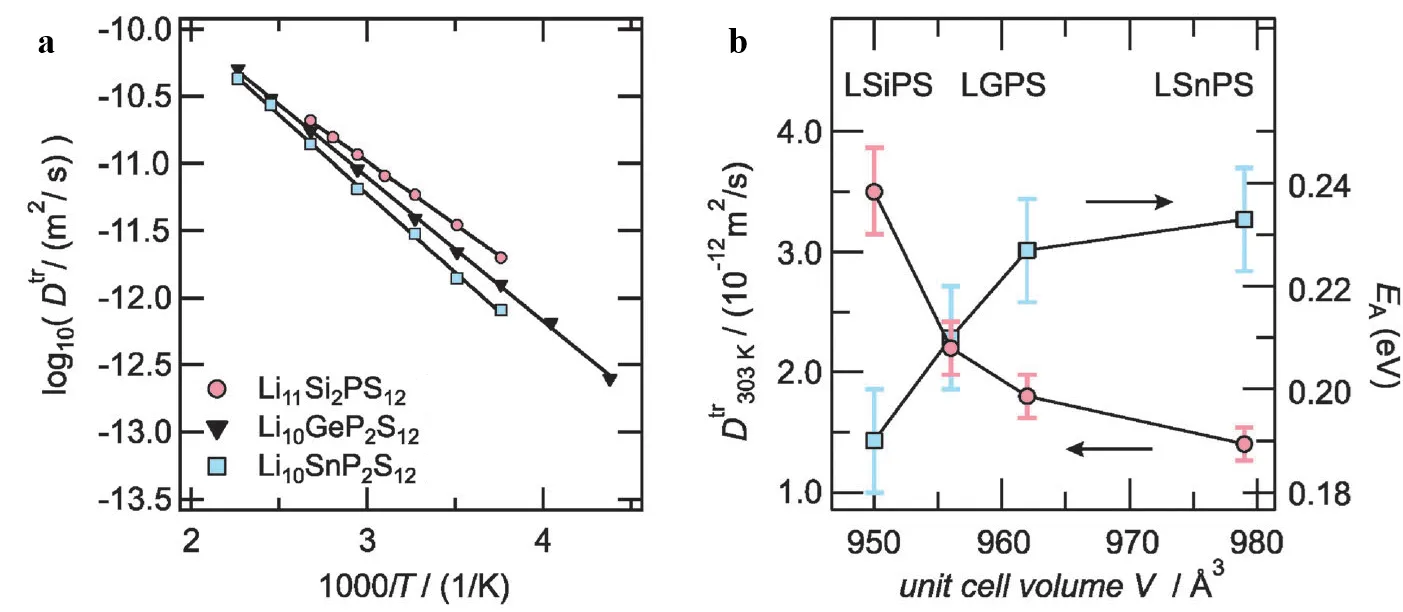
The ionic conductivity of sulfide electrolytes is closely related to their structural characteristics. Taking crystalline Li
10GeP
2S
12 (LGPS) as an example, its unique layered tetragonal structure (space group P4
2/nmc) forms a three-dimensional sulfur skeleton network. Kuhn et al. [
32] found through synchrotron X-ray diffraction and pulsed field gradient nuclear magnetic resonance (PFG-NMR) that Li⁺ ions migrate rapidly mainly through one-dimensional tunnels (Li1–Li3 sites) along the c-axis (diffusion coefficient D = 3.5 × 10
−12 m
2 s
−1, 300 K), while the interstitial octahedral sites (Li4) in the ab plane provide auxiliary paths for ion transport. The synergistic effect of this multidimensional conduction mechanism enables LGPS to exhibit ultra-high ionic conductivity (>12 mS cm
−1). It is worth noting that this study revealed for the first time through ³¹P MAS NMR that Si doping (Li
11Si
2PS
12) can reduce the unit cell volume (a=8.21→8.12 Å, c=6.62→6.59 Å), thereby reducing the activation energy to 0.19 eV, confirming the role of lattice contraction in promoting ion migration, as shown in
b.
. (<strong>a</strong>) Diffusion Coefficients: Measurement of Li tracer diffusion coefficients in LGPS-type electrolytes using <sup>7</sup>Li PFG NMR; (<strong>b</strong>) Diffusion and Volume Correlation: Analysis of the relationship between diffusion parameters and the unit cell volume for LGPS-type electrolytes. The plot shows the variation of diffusion parameters with changes in unit cell volume. Reproduced with permission of Ref. [<a href="#B32" class="html-bibr">32</a>], Copyright 2014 the Owner Societies.
Compared with crystalline materials, the conduction mechanism of the glassy Li
2S−P
2S
5 system shows significant differences. Ooura et al. [
33] prepared an 86.9Li₃PS₄·13.1LiAlS
2 amorphous electrolyte by high-energy ball milling and showed that the addition of Al
2S
3 can effectively reduce the proportion of non-bridging sulfur (NBS). When the Al
2S
3 content increased to 13.1 mol%, the ionic conductivity reached 6.0 × 10
−4 S cm
−1 (298 K), which is 2–3 times higher than that of the undoped Li
3PS
4 system. This improvement is attributed to the formation of a more uniform sulfur network by the [AlS
4]
− tetrahedron and [PS
4]
3− anions sharing vertices, thereby reducing the electrostatic barrier for Li
+ migration. However, excessive doping (>13.1 mol%) leads to Al
2S
3 crystallization, which hinders ion transport.
5.2. Theoretical Calculations to Predict Ion Migration Paths and Migration Energy Barriers
Density functional theory (DFT) in conjunction with the Neural Embedding-Based (NEB) technique constitutes the backbone for exploring the migration paths and energy barriers of ions in solid electrolytes. With an illustration of a common oxide electrolyte, Li
7La
3Zr
2O
12 (LLZO), the NEB technique provides the precise computation of the migration energy barrier of the Li
+ moving between lattice sites [
34,
35]. This technique also exposes its transition state configuration by forming an energy minimization pathway. In recent years, it has gained recent attention for the discovery of the migration pattern of metal ions extended to the study of sulfide antiperovskite systems. Kim et al. [
36] found through systematic DFT calculations that the Li⁺ migration energy barrier and the degree of lattice distortion (quantified by the Goldschmidt tolerance factor t) show a significant negative correlation: in sulfides such as Li
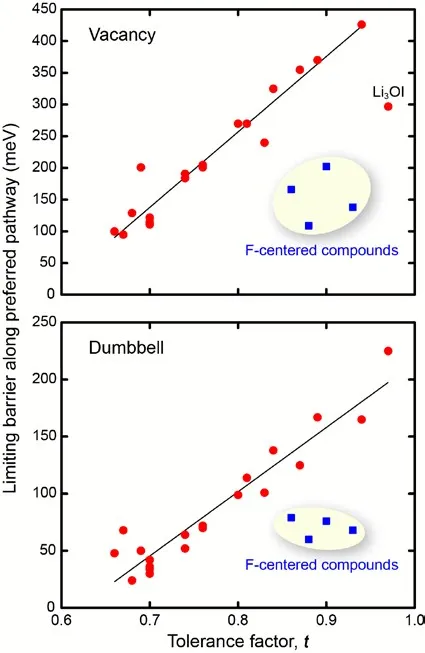
3OCl, the vacancy migration mechanism energy barrier ranges from 95–426 meV, while the interstitial dumbbell mechanism has a significantly reduced energy barrier to 24–165 meV due to the channel widening effect, as shown in
. This discovery shows that by regulating the tolerance factor to induce lattice distortion, specific migration channels can be selectively optimized, providing a theoretical basis for designing high ionic conductivity electrolytes.
. Correlation between the limiting barriers for vacancy (top) and interstitial dumbbell (bottom) migration. Blue squares represent compounds centered at F. Reproduced with permission of Ref. [<a href="#B36" class="html-bibr">36</a>], Copyright 2019 Materials Chemistry A.
AIMD provides a dynamic perspective for ion migration research. Unlike the NEB method, which requires a preset reaction path, AIMD can spontaneously capture the ion migration trajectory and statistically analyze the diffusion characteristics by simulating the dynamic evolution of the system at a finite temperature. Taking Li
10GeP
2S
12 (LGPS) as an example, Mo et al. [
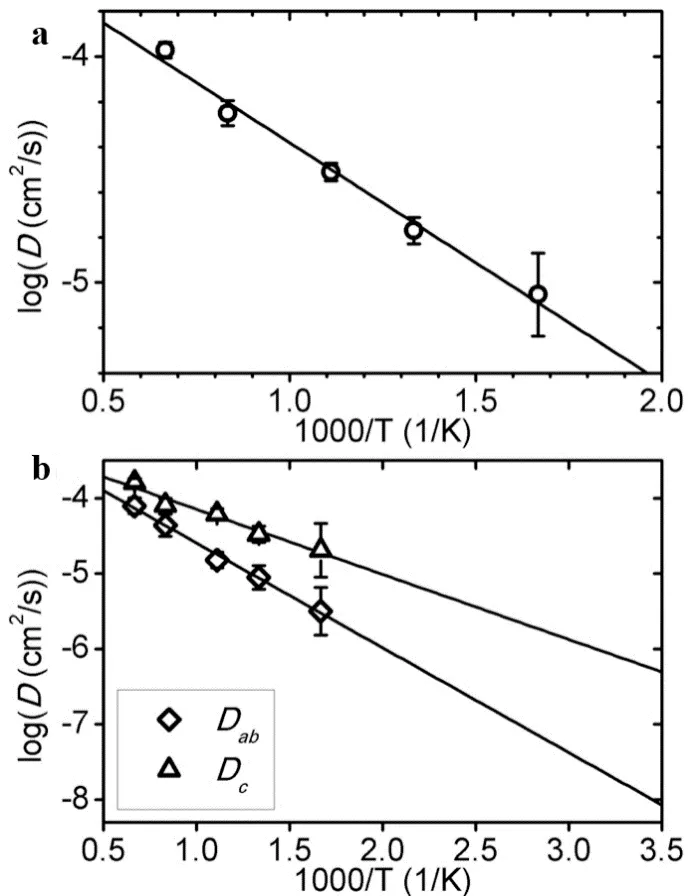
37] revealed its three-dimensional ion conduction network through AIMD simulation (600–1500 K): not only verified the one-dimensional fast conduction channel along the c-axis (activation energy 0.17 eV), but also discovered the cross-channel diffusion path within the ab plane (activation energy 0.28 eV), breaking the traditional belief that LGPS only has one-dimensional conduction characteristics. The calculated overall activation energy (0.21 eV) is highly consistent with the experimental value (0.24 eV), and the predicted range of room temperature ionic conductivity (2–40 mS cm
−1) also covers the experimental measurement value (12 mS cm
−1). The unique advantage of AIMD simulation is that it can capture the cooperative mechanism of ion conduction. By fitting the Arrhenius relationship, not only can the activation energy be obtained, but also the transition of conduction mechanisms in different temperature ranges can be revealed. In LGPS, the conductivity of c-axis conduction at 300 K (40 mS cm
−1) is two orders of magnitude higher than that of ab-plane conduction (0.9 mS cm
−1). However, cross-channel diffusion within the ab plane still makes an important contribution to the overall conduction performance, especially in the presence of grain boundaries or defects. This three-dimensional conduction network can effectively avoid the problem of one-dimensional conduction channels being blocked, as shown in
.
. (<strong>a</strong>) Arrhenius plots illustrating the overall diffusion coefficient, along with the diffusion coefficients in the c-direction (Dc) and the ab-plane (Dab). The plots show the temperature dependence of these coefficients; (<strong>b</strong>) Error bars indicating the statistical uncertainty derived from fitting the mean square displacement to time curves. This provides a measure of the reliability of the diffusion coefficient estimates. Reproduced with permission of Ref. [<a href="#B37" class="html-bibr">37</a>], Copyright 2012 American Chemical Society.
In recent years, MLMD has achieved breakthrough progress in the prediction of ion migration paths by combining neural network potential functions with molecular dynamics, while maintaining the accuracy of first principles and achieving large-scale simulations at the microsecond level. Taking the study of the cooperative diffusion mechanism of Li
10GeP
2S
12 (LGPS) as an example, Fu et al. [
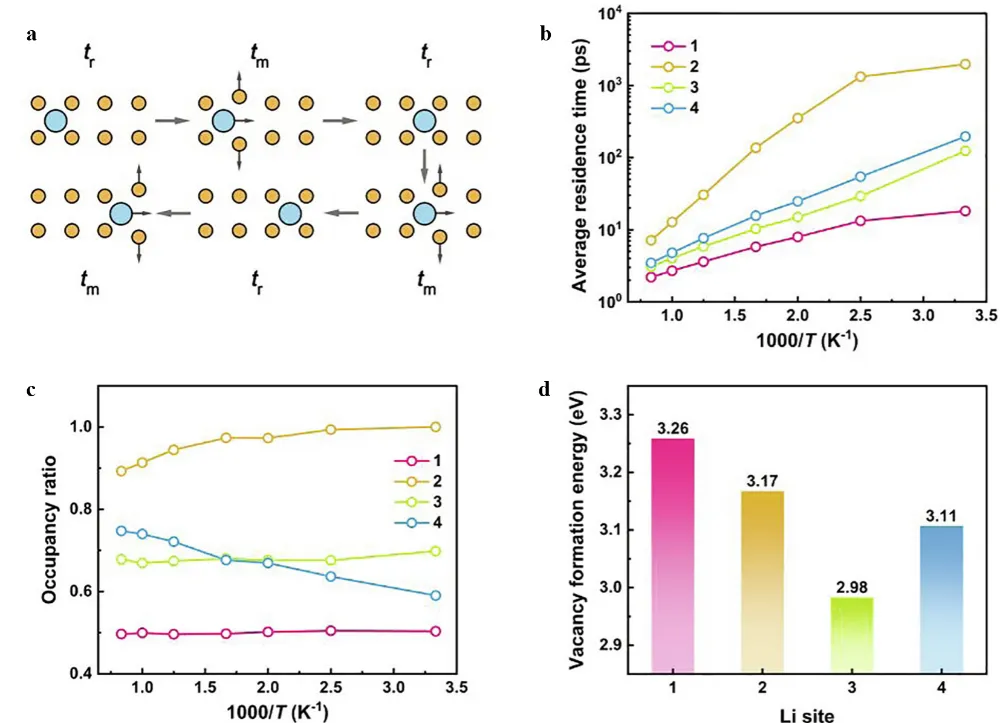
27] captured the temperature-dependent ion migration pattern in a 40 ns long-term simulation by training a deep potential model based on 2400 sets of first principles data, as shown in
. The study found that at 300 K, Li⁺ mainly migrates along the c-axis through three-ion cooperative transitions (Li1–Li3–Li4 mode), with a diffusion coefficient of 7.1 × 10
−12 m
2 s
−1 and an activation energy of 0.23 eV, which is highly consistent with the experimental value (4.1 × 10
−12 m
2 s
−1, 0.24 eV); when the temperature rises to 1000 K, the four-ion cooperative migration involving the two-dimensional Li2 site is activated, and its transition frequency increases linearly with temperature (
R2 = 0.98), causing the diffusion anisotropy index to drop from 0.62 to 0.38. This temperature-induced diffusion mode transition originates from the increase in the vacancy concentration of Li₂ sites (300 K: 0.09→1000 K: 0.21, as shown in
c. Its essence is that the vibration amplitude of the Li-S bond is enlarged (the mean square displacement is from 0.12→0.35 Å
2), which reduces the migration barrier. This work revealed for the first time the Arrhenius temperature-dependent nature of cooperative diffusion through MLMD——the activation energy of different transition modes (0.15–0.36 eV) is weakly correlated with temperature, while the linear increase of the transition frequency dominates the change of macroscopic ionic conductivity.
. Analysis of Li sites in LGPS. (<strong>a</strong>) Schematic representation of the residence time t<sub>r</sub> and migration time t<sub>m</sub> of a mobile ion. (<strong>b</strong>) Average residence time of four Li atoms and (<strong>c</strong>) site occupancy ratio as a function of inverse temperature. Average Li site in the LGPS lattice. (<strong>d</strong>) Li vacancy formation energy. Reproduced with permission of Ref. [<a href="#B27" class="html-bibr">27</a>], Copyright 2022 Elsevier.
6. High-Throughput Screening and Machine Learning
6.1. Applications of High-Throughput Screening
First, construct a candidate material library. In search of new sulfide electrolytes, in high-throughput screening, one can first produce a large number of candidate material structures by building the structure of known sulfides and abiding by the rules of chemical composition of sulfides. For example, if one changes the metal type and proportion of the elements of the sulfide or inserts other elements for doping, a candidate sulfide electrolyte material structure of hundreds of different structures and compositions can be constructed,
etc. These types of candidate materials can represent various stoichiometric ratios and crystal structure types, thus affording a wealth of structural examples for subsequent searching. With the assistance of automatically written scripts and computational chemistry software, the primitive structural models of candidate materials, such as atom coordinates, lattice parameters and others, could be rapidly generated so that they would be ready for subsequent searching in terms of material stability and calculation in terms of material performance.
Secondly, stability analysis is performed. High-throughput screening can rapidly screen the thermodynamic stability of candidate sulfide electrolytes. Calculating the formation energy of each candidate material, the stability under specific conditions can be determined. By matching the formation energy of a large number of candidate sulfide electrolytes through the first-principles calculations (such as DFT) together with a high-throughput computing framework, the thermodynamic stability of sulfide electrolytes can be predicted by calculating the formation energy of sulfide electrolytes in batches if it is small, which is difficult to decompose.
At the same time, it is possible to estimate its kinetic stability, for example, the change of structure of candidate materials in a simulated electrochemical environment by molecular dynamics simulation. As long as the structure of the material in the simulation does not collapse or undergo any phase transition, then the material has good kinetic stability in the work application.
6.2. Performance Prediction: High-Throughput Calculation from Ion Migration to Electrochemical Stability
In the performance prediction of sulfide electrolytes, the coordinated optimization of ionic conductivity and electrochemical stability window is the core goal. For ion migration characteristics, the NEB method based on DFT can accurately calculate the migration energy barrier of lithium/sodium ions by constructing a multi-image reaction path [
37]. In the study of adsorbed atom diffusion on the Cu (100) surface, Jónsson et al. [
38,
39] verified the consistency of the migration energy barrier with the experimental value by the NEB method (error < 0.02 eV), and pointed out that the system with an energy barrier below 0.3 eV can achieve a room temperature ionic conductivity of >10
−3 S cm
−1. Combined with a high-throughput computing framework, this method can batch evaluate the topological characteristics of the migration path of candidate materials (such as cooperative migration or vacancy-dominated mechanism), significantly improving the screening efficiency.
For the prediction of the electrochemical stability window, it is necessary to combine electronic structure analysis with thermodynamic phase equilibrium calculation. The traditional HOMO-LUMO method estimates the upper limit of the stability window by band gap, as shown in
, but it ignores the interface dipole effect, resulting in an absolute potential prediction error of up to 1.0 V [
40]. Recent studies have proposed a three-dimensional phase diagram method (Phase Stability Method) based on Gibbs free energy. By calculating the equilibrium potential of the decomposition reaction Li
xM→Li
x − ΔM + ΔLi, the stability window boundary can be more accurately defined. Taking Li
7La
3Zr
2O
12 (LLZO) as an example, the HSE06 hybrid functional calculation shows that its phase stability window is 0.0–2.84 V vs. Li⁺/Li, which is 0.68 V wider than the PBE functional prediction value, highlighting the influence of the exchange correlation functional selection on the prediction accuracy as shown in
.
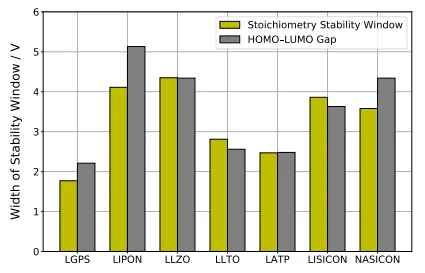
. The width ∆Φ<sup>stoi</sup> of the stoichiometric stability window of the studied SSE materials compared to the HOMO-LUMO gap $$\Delta \mathrm{E}_{\mathrm{H L}}^{\mathrm{N}}$$. Reproduced with permission of Ref. [<a href="#B40" class="html-bibr">40</a>], Copyright 2019 Royal Society of Chemistry.
.
Computational Results from Hybrid Functional DFT Using HSE06 Functional and SG15 ONCV Pseudopotentials: The table presents the lower and upper limits ($$\Phi_{\mathrm{r e d}}^{\mathrm{s t o i}} $$ and $$\Phi_{\mathrm{o x}}^{\mathrm{s t o i}}$$) of the stoichiometric stability windows, the corresponding width (∆Φstoi), and the electronic HOMO–LUMO gaps ($$\Delta \mathrm{E}_{\mathrm{H L}}^{\mathrm{N}}$$) for systems with stable A-stoichiometry N. Reproduced with permission of Ref. [40], Copyright 2019 Royal Society of Chemistry.
| Material |
$$\mathbf{\Phi}_{\mathbf{r} \mathbf{e} \mathbf{d}}^{\mathbf{s} \mathbf{t} \mathbf{o} \mathbf{i}}$$/Vvs.Li |
$$\mathbf{\Phi}_{\mathbf{o} \mathbf{x}}^{\mathbf{s} \mathbf{t} \mathbf{o} \mathbf{i}}$$/Vvs.Li |
∆Φstoi/V |
$$\mathbf{\Delta} \mathbf{E}_{\mathbf{H} \mathbf{L}}^{\mathbf{N}}$$/eV |
| LLZO |
−1.08 |
3.92 |
5.00 |
5.81 |
| LLTO |
1.44 |
5.36 |
3.92 |
4.28 |
| LATP |
0.75 |
4.03 |
3.28 |
4.19 |
6.3. Discovery of Novel Sulfide Electrolytes by High-Throughput Screening
High-throughput screening technology based on multi-scale computing has become a key paradigm for discovering high-performance solid-state electrolytes. Taking the discovery of the sulfur-based superionic conductor Li
10GeP
2S
12(LGPS) as an example, the research team systematically analyzed the thermodynamic stability and migration path topological characteristics of the Li-Ge-PS quaternary system by constructing a multidimensional screening framework of “chemical composition-crystal structure-ion transport characteristics”. Fu et al. [
27] used MLMD and deep potential energy models to reveal for the first time the temperature-dependent cooperative diffusion mechanism of lithium ions in LGPS: in the range of 300–1200 K, the cooperative migration of Li
+ through the Li1-Li3-Li1 chain channel showed Arrhenius-type temperature dependence, and its activation energy was only 0.23 eV, as shown in
e. This potential energy surface smoothing effect caused by the cooperative motion of multiple ions enables LGPS to obtain an ultra-high ionic conductivity of 12 mS·cm
−1 at room temperature, which is two orders of magnitude higher than traditional oxide electrolytes. More importantly, through dynamic site occupancy analysis, this study found that the activation of Li2 sites under high temperature conditions significantly enhanced the ab plane diffusion contribution, as shown in
b–d, providing a new idea for the anisotropic regulation of sulfide electrolytes.
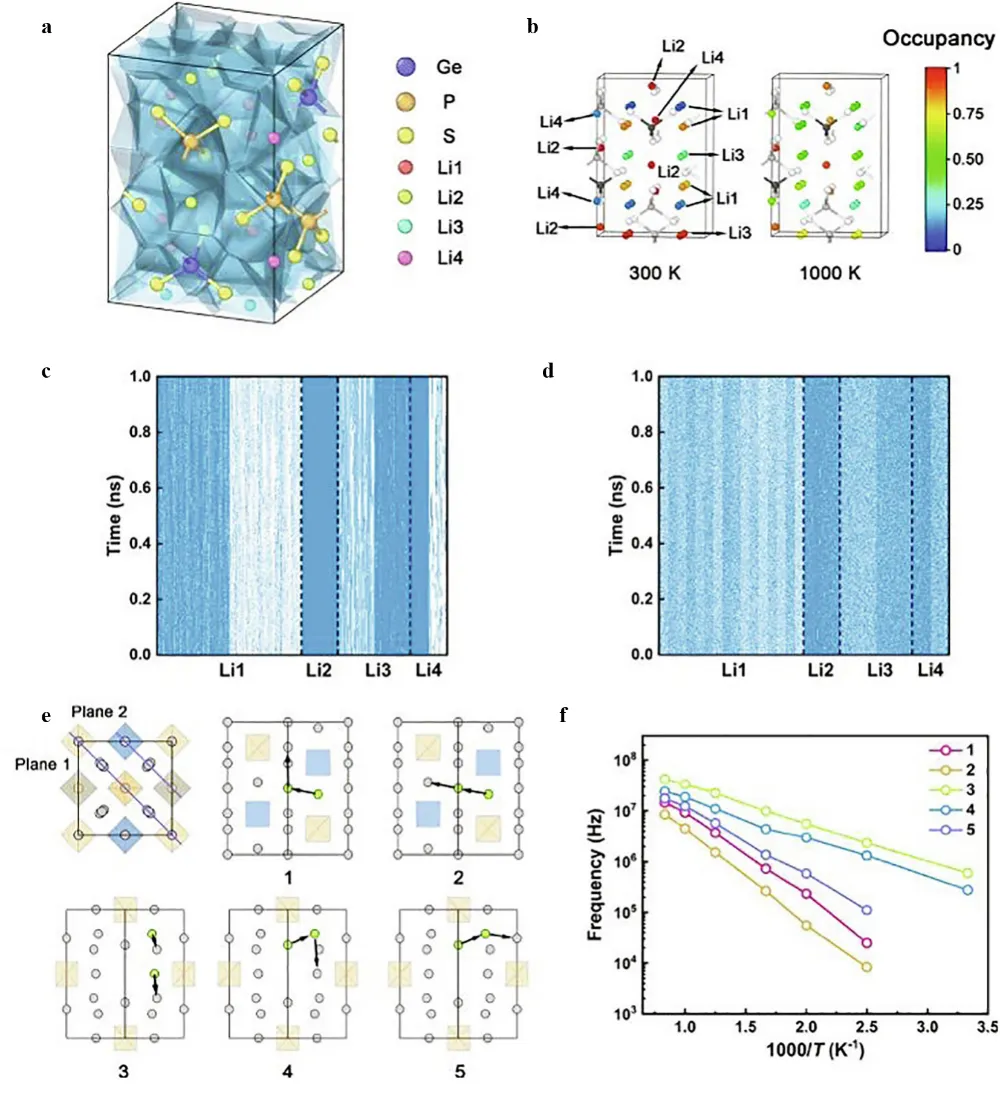
. The diffusion mechanism in LGPS. (<b>a</b>) Voronoi tessellation visualization of the LGPS crystal structure with different lithium (Li) sites; (<b>b</b>) Visualization of lithium site occupation in LGPS at two temperature points, 300 K and 1000K, displaying the influence of temperature on the lithium concentration.Spatial distribution of the time evolution of Li site occupancy at (<b>c</b>) 300 K and (<b>d</b>) 1000 K, the blue (white) grid points denote occupied (unoccupied) state respectively; (<b>e</b>) Schematic representation of the possible cooperative pathways for diffusion on the (110) plane of LGPS, plane 1 and Plane 2 include pathways 1–2 and 3–5, respectively. The relative orientation of these planes is shown in the inset; (<b>f</b>) Degrade of the hopping frequency of the cooperative diffusion mode with temperature. Reproduced with permission of Ref. [
27], Copyright 2022 Elsevier.
In the study of doping optimization of LLZO (Li
7La
3Zr
2O
12) electrolytes, machine learning technology provides an efficient prediction tool for material design by establishing a complex mapping relationship between element doping and ionic conductivity. For example, based on random forest or deep neural network models, researchers can build a high-precision prediction system by integrating experimental and computational data such as doping element type, concentration, lattice parameters and corresponding ionic conductivity [
41]. After the model training is completed, the candidate doping scheme can be input to quickly screen out potential high-performance combinations. This method is particularly suitable for complex systems with synergistic effects of multiple variables. Gao et al. [
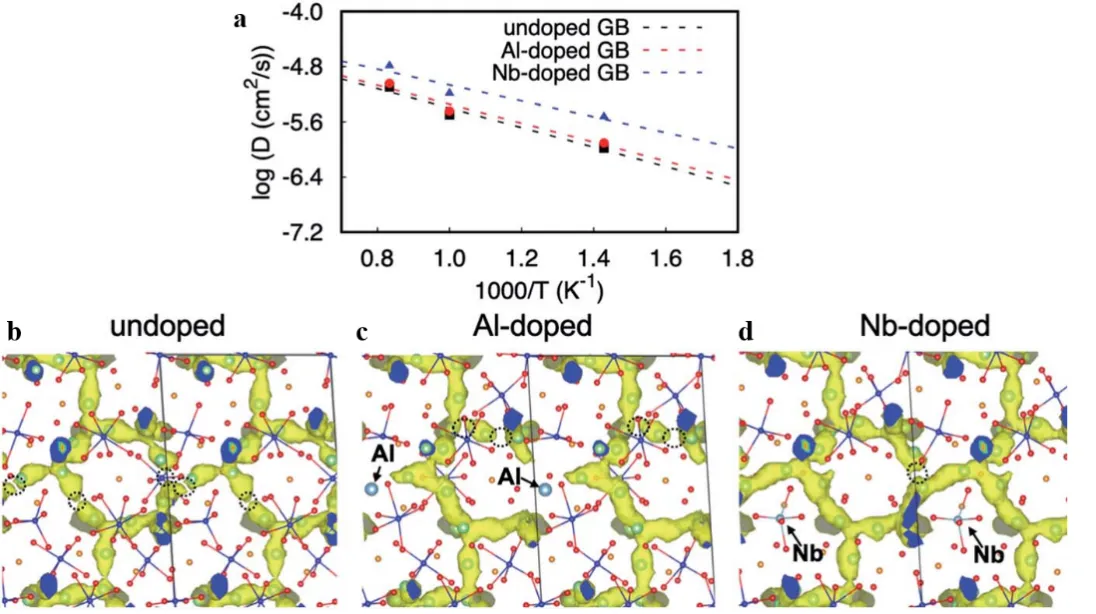
42] revealed the key role of doping atoms in LLZO grain boundaries (GBs) through AIMD simulations. The study found that Nb dopants significantly improved the ionic conductivity of the grain boundary region (σ(300 K) reached 2.66 × 10
−3 S cm
−1) by reducing the Li concentration and increasing the Li vacancy density, while Al doping hindered the Li⁺ migration network, resulting in a conductivity similar to that of the undoped system (3.85 × 10
−4 S cm
−1). Based on such atomic-scale mechanisms, machine learning models can predict the nonlinear relationship between Nb doping ratio and conductivity gain. Subsequent experimental verification showed that when the Nb doping concentration is 0.15–0.32 mol%, the bulk ionic conductivity of LLZO can be increased to the order of 10
−3 S cm
−1, which is highly consistent with the simulation prediction trend, as shown in
.
. (<b>a</b>) Arrhenius plots showing the diffusion coefficients for undoped, Al-doped, and Nb-doped S3(112) grain boundary (GB) models of LLZO. The plots illustrate the temperature dependence of diffusion in these systems; (<b>b</b>–<b>d</b>) Partial Li-ion trajectory densities accumulated from 20 to 55 ps in FPMD simulations at 1000 K for (<b>b</b>) undoped, (<b>c</b>) Al-doped, and (<b>d</b>) Nb-doped S3(112) GB models. The dashed circle highlights the disconnection in the trajectory density. In (<b>c</b>) and (<b>d</b>), light blue spheres indicate the positions of Al and Nb dopants, respectively, with arrows marking their locations. Reproduced with permission of Ref. [
42], Copyright 2022 The Royal Society of Chemistry.
Based on the reverse design idea of machine learning, researchers set target performance requirements, such as hoping to obtain a sulfide electrolyte with an ionic conductivity higher than a certain value and a wide electrochemical window width. Then, the machine learning model is used to reversely analyze possible material feature combinations, such as specific element ratio ranges, crystal structure parameters,
etc. [
43]. Based on these feature combinations, new sulfide electrolyte structures are designed. After high-throughput screening and experimental verification, it was found that these newly designed sulfide electrolytes did have significant improvements in performance and met the application requirements of high-performance electrolytes. Chen et al. [
44] used a machine learning-assisted hierarchical screening strategy to quickly and accurately identify sulfide electrolyte candidate materials with high ionic conductivity from 20,717 lithium-containing materials. They used machine learning models to predict the ionic conductivity of the materials and combined AIMD simulations to verify the ion transport performance of these materials. Finally, they discovered three new sulfide electrolytes with high room temperature ionic conductivity: Li
3BiS
3, Li
5BiS
4, and Li
10ZnP
4S
16. In addition, Zhang et al. [
45] used machine learning models, combined with high-throughput computing and experimental verification, to design new sulfide electrolytes. They predicted the ionic conductivity of the materials through machine learning models and validated the performance of these materials through experiments. The test results showed that the newly designed sulfide electrolytes exhibited good performance, which was suitable for high-performance electrolytes’ application requirements.
7. Conclusions and Outlook
Sulfide solid electrolyte has become one of the main focuses of the whole-field of research of all-solid-state batteries owing to its high ionic conductivity and broad temperature adaptability. Based on the theoretical calculation and simulation, a deep explanation has been made for the ion transport path, activation energy and interface reaction mechanism of sulfide electrolytes, and the design of element doping and structural optimization, and interface engineering has been directed. For instance, the oxygen doping drastically enhances the electrochemical stability of the sulfide electrolytes, and the LiNbO3 buffer layer can effectively avoid the interfacial side reactions. The integrations of machine learning and high-throughput screening promote the development of high-performance materials (e.g., Li10GeP2S12) and suggest new doping methods (e.g., Nb-doped LLZO). However, the application of sulfide electrolytes still has some difficulties: the first one is the narrow electrochemical window which restricts its compatibleness with high-voltage positive electrodes; the second one is the impedance problem which caused by bad solid-solid interface contact has not been overcome; the third one is that sulfides are hygroscopic, and big-scale preparation process urgently requires breaking through. The future research can be done along the following directions: (1) Research new sulfide system containing high ionic conductivity and wide voltage window; (2) Integration of multi-scale simulations and experiment to devise composite interface layer that are chemically and mechanically stable; (3) Study the atmospheric synthesis process of sulfide electrolyte to achieve the low cost effect. Moreover, material reverse design driven by artificial intelligence should also breach the traditional empirical trial and error model and open up new concepts for all-solid-state battery practical application.
Acknowledgments
This work was supported by Guangdong Province NaturaScience Foundation (2023A1515011122) and South China Normal University Extracurricular Research General Project Cultivation Program (24XZGB07). Youth Teacher Research and Cultivation Fund of South China Normal University (Project No. 671866), presided over, 30,000 CNY.
Author Contributions
Conceptualization, Y.F.; Methodology, J.L. and Z.Z.; Validation, Y.F., J.L. and Y.H.; Formal Analysis, G.H. and L.L.; Investigation, Y.F., Z.W. and H.H.; Resources, J.L.; Data Curation, H.H. and G.H.; Writing-Original Draft Preparation, Y.F.; Writing-Review & Editing, J.L., Z.Z. and J.C.; Visualization, Z.L. and G.H; Supervision, J.Z. and L.Z.; Project Administration, J.L. and Y.H.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable to this article, as no new data were created in this study.
Funding
This research was funded by Guangdong Province NaturaScience Foundation (2023A1515011122); South China Normal University Extracurricular Research General Project Cultivation Program (24XZGB07) and Youth Teacher Research and Cultivation Fund of South China Normal University (Project No. 671866).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AI Statement
During the preparation of this work, the authors used Deepseek in order to improve readability and language. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication. Explanation of the use of AI. 1. Since my learning ability is still insufficient, and I do not express and summarize the literature data well, thus, I use AI tools to make the data presentation more accurate. 2. In the process of writing the article, because I need to use another language, and in order to better present the article, I used AI to help me correct the grammar of the sentence and the smoothness of the sentence.
References
-
1.
Manthiram A, Yu X, Wang S. Lithium battery chemistries enabled by solid-state electrolytes.
Nat. Rev. Mater. 2017,
2, 16103.
[Google Scholar]
-
2.
Kamaya N, Homma K, Yamakawa Y, Hirayama M, Kanno R, Yonemura M, et al. A lithium superionic conductor.
Nat. Mater. 2011,
10, 682–686.
[Google Scholar]
-
3.
Kato Y, Hori S, Saito T, Suzuki K, Hirayama M, Mitsui A, et al. High-power all-solid-state batteries using sulfide superionic conductors.
Nat. Energy 2016,
1, 16030.
[Google Scholar]
-
4.
Seino Y, Ota T, Takada K, Hayashi A, Tatsumisago M. A sulphide lithium super ion conductor is superior to liquid ion conductors for use in rechargeable batteries.
Energy Environ. Sci. 2014,
7, 627–631.
[Google Scholar]
-
5.
Lee D, Kumar Kakarla A, Sun S, Joohyun Kim P, Choi J. Inorganic Solid-State Electrolytes for Solid-State Sodium Batteries: Electrolyte Design and Interfacial Challenges.
Chemelectrochem 2025,
12, e202400612.
[Google Scholar]
-
6.
Hayashi A, Hama S, Minami T, Tatsumisago M. Formation of superionic crystals from mechanically milled Li2S–P2S5 glasses.
Electrochem. Commun. 2003,
5, 111–114.
[Google Scholar]
-
7.
Deiseroth HJ, Kong ST, Eckert H, Vannahme J, Reiner C, Zaiß T, et al. Li6PS5X: A Class of Crystalline Li-Rich Solids With an Unusually High Li+ Mobility
Angew. Chem. Int. Ed. 2008,
47, 755.
[Google Scholar]
-
8.
Zhu Y, He X, Mo Y. Origin of outstanding stability in the lithium solid electrolyte materials: insights from thermodynamic analyses based on first-principles calculations.
ACS Appl. Mater. Interfaces 2015,
7, 23685–23693.
[Google Scholar]
-
9.
Komatsu H, Banerjee S, Holekevi Chandrappa ML, Qi J, Radhakrishnan B, Kuwata S, et al. Interfacial Stability of Layered LiNi
xMn
yCo
1–x–yO
2 Cathodes with Sulfide Solid Electrolytes in All-Solid-State Rechargeable Lithium-Ion Batteries from First-Principles Calculations.
J. Phys. Chem. C 2022,
126, 17482–17489.
[Google Scholar]
-
10.
Wan S, Zhao S, Ma W, Chen S. Computational approaches to electrolyte design for advanced lithium-ion batteries.
Chem. Commun. 2025,
61, 7019–7034. doi:10.1039/d5cc01310k.
[Google Scholar]
-
11.
Yu S, Siegel DJ. Grain boundary contributions to Li-ion transport in the solid electrolyte Li
7La
3Zr
2O
12 (LLZO).
Chem. Mater. 2017,
29, 9639–9647.
[Google Scholar]
-
12.
He X, Zhu Y, Epstein A, Mo Y. Statistical variances of diffusional properties from ab initio molecular dynamics simulations.
Npj. Comput. Mater. 2018,
4, 18.
[Google Scholar]
-
13.
Haruyama J, Sodeyama K, Tateyama Y. Cation mixing properties toward Co diffusion at the LiCoO2 cathode/sulfide electrolyte interface in a solid-state battery.
ACS Appl. Mater. Interfaces 2017,
9, 286–292.
[Google Scholar]
-
14.
Camacho-Forero LE, Balbuena PB. Exploring interfacial stability of solid-state electrolytes at the lithium-metal anode surface.
J. Power Source 2018,
396, 782–790.
[Google Scholar]
-
15.
Takada K, Ohta N, Zhang L, Xu X, Hang BT, Ohnishi T, et al. Interfacial phenomena in solid-state lithium battery with sulfide solid electrolyte.
Solid State Ion. 2012,
225, 594–597.
[Google Scholar]
-
16.
Koerver R, Aygün I, Leichtweiß T, Dietrich C, Zhang W, Binder JO, et al. Capacity fade in solid-state batteries: interphase formation and chemomechanical processes in nickel-rich layered oxide cathodes and lithium thiophosphate solid electrolytes.
Chem. Mater. 2017,
29, 5574–5582.
[Google Scholar]
-
17.
Xu H, Yu Y, Wang Z, Shao G. A theoretical approach to address interfacial problems in all-solid-state lithium ion batteries: tuning materials chemistry for electrolyte and buffer coatings based on Li6PA5 Cl hali-chalcogenides.
J. Mater. Chem. A 2019,
7, 5239–5247.
[Google Scholar]
-
18.
Yamagishi Y, Morita H, Nomura Y, Igaki E. Visualizing lithium distribution and degradation of composite electrodes in sulfide-based all-solid-state batteries using operando time-of-flight secondary ion mass spectrometry.
Acs. Appl. Mater. Interfaces 2020,
13, 580–586.
[Google Scholar]
-
19.
Auvergniot J, Cassel A, Foix D, Viallet V, Seznec V, Dedryvère R. Redox activity of argyrodite Li
6PS
5Cl electrolyte in all-solid-state Li-ion battery: An XPS study.
Solid State Ion. 2017,
300, 78–85.
[Google Scholar]
-
20.
Sakuda A, Hayashi A, Tatsumisago M. Interfacial observation between LiCoO
2 electrode and Li2S−P2S5 solid electrolytes of all-solid-state lithium secondary batteries using transmission electron microscopy.
Chem. Mater. 2010,
22, 949–956.
[Google Scholar]
-
21.
Richards WD, Miara LJ, Wang Y, Kim JC, Ceder G. Interface stability in solid-state batteries.
Chem. Mater. 2016,
28, 266–273.
[Google Scholar]
-
22.
Bartók AP, Csányi G. G aussian approximation potentials: A brief tutorial introduction.
Int. J. Quantum Chem. 2015,
115, 1051–1057.
[Google Scholar]
-
23.
Oh K, Chang D, Lee B, Kim D, Yoon G, Park I, et al. Native defects in Li
10GeP
2S
12 and their effect on lithium diffusion.
Chem. Mater. 2018,
30, 4995–5004.
[Google Scholar]
-
24.
Behler J. Four generations of high-dimensional neural network potentials.
Chem. Rev. 2021,
121, 10037–10072.
[Google Scholar]
-
25.
Pun GP, Batra R, Ramprasad R, Mishin Y. Physically informed artificial neural networks for atomistic modeling of materials.
Nat. Commun. 2019,
10, 2339.
[Google Scholar]
-
26.
Wang H, Zhang L, Han J. DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics.
Comput. Phys. Commun. 2018,
228, 178–184.
[Google Scholar]
-
27.
Fu Z-H, Chen X, Yao N, Shen X, Ma X-X, Feng S, et al. The chemical origin of temperature-dependent lithium-ion concerted diffusion in sulfide solid electrolyte Li10GeP2S12
J. Energy Chem. 2022,
70, 59–66.
[Google Scholar]
-
28.
Zhang Z, Ma Z, Pei Y. Li ion diffusion behavior of Li
3OCl solid-state electrolytes with different defect structures: insights from the deep potential model.
Phys. Chem. Chem. Phys. 2023,
25, 13297–13307.
[Google Scholar]
-
29.
Li H, Zhou X, Wang Y, Jiang H. Theoretical study of Na+ transport in the solid-state electrolyte Na
3OBr based on deep potential molecular dynamics.
Inorg. Chem. Front 2021,
8, 425–432.
[Google Scholar]
-
30.
Wang S, Liu Y, Mo Y. Frustration in super-ionic conductors unraveled by the density of atomistic states.
Angew. Chem. Int. Ed. 2023,
62, e202215544.
[Google Scholar]
-
31.
Xu Z, Duan H, Dou Z, Zheng M, Lin Y, Xia Y, et al. Machine learning molecular dynamics simulation identifying weakly negative effect of polyanion rotation on Li-ion migration.
Npj Comput. Mater. 2023,
9, 105.
[Google Scholar]
-
32.
Kuhn A, Gerbig O, Zhu C, Falkenberg F, Maier J, Lotsch BV. A new ultrafast superionic Li-conductor: ion dynamics in Li
11Si
2PS
12 and comparison with other tetragonal LGPS-type electrolytes.
Phys. Chem. Chem. Phys. 2014,
16, 14669–14674.
[Google Scholar]
-
33.
Ooura Y, Machida N, Naito M, Shigematsu T. Electrochemical properties of the amorphous solid electrolytes in the system Li2S–Al2S3–P2S5.
Solid State Ion. 2012,
225, 350–353.
[Google Scholar]
-
34.
Maiti S, Curnan MT, Maiti K, Choung S, Han JW. Accelerating Li-based battery design by computationally engineering materials.
Chem 2023,
9, 3415–3460.
[Google Scholar]
-
35.
Sradhasagar S, Pati S, Roy A. Computational Design of an Affordable, Lightweight Solid Electrolyte for All-Solid-State Lithium-Ion Batteries.
J. Phys. Chem. C 2024,
128, 15242–15254.
[Google Scholar]
-
36.
Kim K, Siegel DJ. Correlating lattice distortions, ion migration barriers, and stability in solid electrolytes.
J. Mater. Chem. A 2019,
7, 3216–3227.
[Google Scholar]
-
37.
Mo Y, Ong SP, Ceder G. First principles study of the Li10GeP2S12 lithium super ionic conductor material.
Chem. Mater. 2012,
24, 15–17.
[Google Scholar]
-
38.
Henkelman G, Jónsson H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points.
J. Chem. Phys. 2000,
113, 9978–9985.
[Google Scholar]
-
39.
Jónsson H, Mills G, Jacobsen KW. Classical and Quantum Dynamics in Condensed Phase Simulations; World Scientific: Singapore, 1998; pp. 385–404.
-
40.
Binninger T, Marcolongo A, Mottet M, Weber V, Laino T. Comparison of computational methods for the electrochemical stability window of solid-state electrolyte materials.
J. Mater. Chem. A 2020,
8, 1347–1359.
[Google Scholar]
-
41.
Wang H, Ji Y, Li Y. Simulation and design of energy materials accelerated by machine learning.
WIREs Comput. Mol. Sci. 2020,
10, e1421.
[Google Scholar]
-
42.
Gao B, Jalem R, Tateyama Y. Atomistic insight into the dopant impacts at the garnet Li7La3Zr2O12 solid electrolyte grain boundaries
J. Mater. Chem. A 2022,
10, 10083–10091.
[Google Scholar]
-
43.
Schaarschmidt J, Yuan J, Strunk T, Kondov I, Huber SP, Pizzi G, et al. Workflow engineering in materials design within the battery 2030+ project.
Adv. Energy Mater. 2022,
12, 2102638.
[Google Scholar]
-
44.
Chen W, Zhou J, Li S, Lu C, Li H, Li Y, et al. Accelerated discovery of novel inorganic solid-state electrolytes through machine learning-assisted hierarchical screening.
J. Alloys Compd. 2025,
1010, 177981.
[Google Scholar]
-
45.
Zhang Y, He X, Chen Z, Bai Q, Nolan AM, Roberts CA, et al. Unsupervised discovery of solid-state lithium ion conductors.
Nat. Commun. 2019,
10, 5260.
[Google Scholar]