1. Introduction
As petroleum reserves continue to dwindle and environmental issues become increasingly severe, efforts are being made to expand and innovate within the traditional petrochemical industry while seeking new primary carbon sources to meet the demands of chemical production. Methane, which is abundant and cost-effective, is widely found in natural gas, coalbed methane, shale gas, and combustible ice, making it a promising alternative to the depleting petroleum resources [
1,
2,
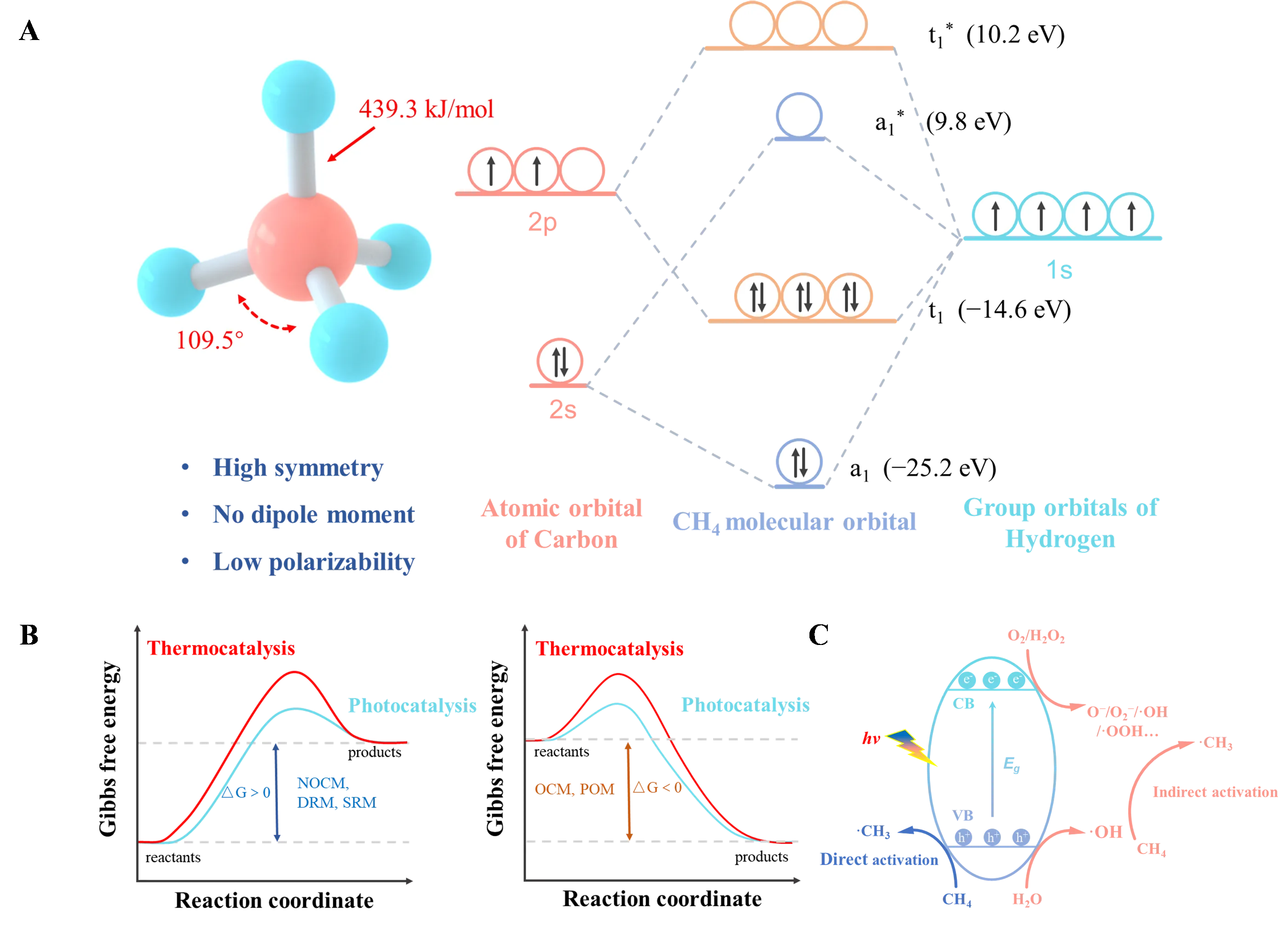
3]. However, as shown in A, the methane molecule is a tetrahedral structure with a bond angle of 109.5°, exhibiting high symmetry and low polarizability. It is a typical non-polar molecule, with a C—H bond dissociation energy of 439 kJ·mol
−1. The molecular orbitals of methane reveal a very low energy for the highest occupied molecular orbital (HOMO) and a very high energy for the lowest unoccupied molecular orbital (LUMO), indicating that both donating and accepting an electron require substantial energy, making methane exceedingly difficult to activate [
4]. Additionally, most methane reserves are located in remote areas, and the transportation and storage of gaseous methane are costly and prone to leakage, potentially causing significant environmental problems [
5,
6]. Therefore, converting methane into transportable and higher-value chemicals is a promising yet challenging approach [
7].
Methane conversion can be categorized into two main types: direct and indirect conversion [
8]. The indirect conversion of methane, also known as the syngas (H
2 and CO) route [
9,
10], involves reforming methane to syngas, which is then further converted into high-value chemical feedstocks such as ammonia, methanol, and olefins [
11]. This is currently the primary commercial route for large-scale methane conversion [
12,
13,
14]. However, indirect conversion methods such as dry reforming of methane (DRM) and steam reforming of methane (SRM) are thermodynamically unfavorable at room temperature ( and B), requiring high temperatures and pressures, resulting in high energy consumption and costs, which are detrimental to sustainable development. Compared to these methane reforming reactions, partial oxidation of methane (POM) to syngas is a spontaneous reaction that can reduce energy consumption and produce a H
2/CO ratio of 2/1, which is more suitable for subsequent processes such as methanol synthesis and Fischer-Tropsch synthesis. Direct conversion of methane involves converting methane directly into high-value chemicals, such as liquid oxygenates and hydrocarbons [
15,
16]. Compared to indirect conversion, direct conversion bypasses intermediate steps, saving costs and utilizing methane more efficiently. Direct conversion of methane includes oxidative and non-oxidative pathways. The non-oxidative pathway, or non-oxidative coupling of methane (NOCM), is an endothermic reaction typically requiring extremely high temperatures (>1000 °C) to activate the inert C—H bond [
17], which can easily cause catalyst deactivation due to carbon deposition [
18]. Generally, introducing oxidants (e.g., O
2, H
2O
2) can effectively lower the Gibbs free energy of the reaction ( and B), allowing the reaction to proceed under milder conditions. Oxidative pathways such as partial oxidation of methane (POM) and oxidative coupling of methane (OCM) are of particular interest due to their low energy consumption and good atom economy. Despite this, in industrial applications of thermal catalytic direct conversion of methane, it remains challenging to couple methane into C
2+ hydrocarbons at very low temperatures and avoid the formation of a large amount of thermodynamically favored by-products such as CO
2 [
19]. Partial oxidation of methane also struggles to selectively oxidize methane into oxygenates and typically requires expensive oxidants such as H
2O
2 [
20].
. (<b>A</b>) Structure (left) and molecular orbital diagrams (right) of methane. * represents the antibonding orbitals. (<b>B</b>) Thermodynamically unfavourable (ΔG > 0) (left) and favourable (ΔG < 0) (right) reaction pathways for methane conversion operated at room temperature. (<b>C</b>) Schematic diagram of direct and indirect methane activation over a semiconductor photocatalyst.
. Change of Gibbs free energy for various methane conversion reactions.
Photocatalysis, as an emerging technology, utilizes widely available and environmentally friendly solar energy to activate alkane’s inert C—H bond under mild conditions [
21,
22,
23]. As illustrated in B, the introduction of photon energy significantly lowers the activation barrier for methane, breaking the thermodynamic equilibrium, which is crucial for achieving efficient low-temperature methane conversion [
24,
25,
26,
27,
28]. Since the initial report by Kaliaguine et al. in 1978 on the photocatalytic conversion of methane, where CH
3O
− and C
2H
5O
− species were observed on TiO
2 surfaces under UV irradiation [
29], photocatalytic aerobic conversion of methane has garnered increasing attention. This process can be divided into two pathways: direct activation and indirect activation of methane [
30]. As depicted in C, when light irradiates the surface of a photocatalyst, photons with energy greater than the semiconductor band gap are absorbed, causing electrons to transition from the valence band (VB) to the conduction band (CB), leaving holes in the VB. The photogenerated electrons and holes migrate to the catalyst surface, where they can react with additional reactants such as H
2O, O
2, and H
2O
2 to generate reactive oxygen species (·OH, ·OOH, ·O
2−), which assist in methane activation (indirect activation pathway). Photogenerated holes can also accumulate on lattice oxygen, forming O
−, thereby directly activating methane to generate ·CH
3 (direct activation pathway). Activated methane can then couple to form C
2+ hydrocarbons (POCM) or combine with reactive oxygen species to form oxygenates (PPOM). Commonly used oxidants include H
2O
2, H
2O, and O
2. H
2O
2 is expensive, difficult to store and transport, and prone to decomposition under high pressure and heating, making it unsuitable for industrial applications. H
2O has low reactivity and is thermodynamically unfavorable (). In contrast, O
2 is economical, environmentally friendly, and thermodynamically favorable, providing unique advantages in photocatalytic aerobic conversion [
31,
32]. However, the complex nature of the reactive oxygen species generated by O
2 presents a significant challenge in controlling product selectivity for researchers.
This review summarizes recent advances in the photocatalytic conversion of methane using O
2 as the oxidant, focusing on the photocatalytic partial oxidation of methane (PPOM) and the oxidative coupling of methane (POCM). The reaction mechanisms in various systems, including C—H bond activation, O
2 reduction, and radical intermediate pathways, are examined, followed by an introduction to photochemical cycling strategies. Finally, the challenges and future prospects in the field of photocatalytic aerobic conversion of methane are discussed.
2. Photocatalytic Partial Oxidation of Methane (PPOM)
Total oxidation of methane produces low value CO
2, a greenhouse gas with adverse environmental impacts, making it unsustainable. In contrast, photocatalytic partial oxidation of methane (PPOM) can proceed under mild conditions to yield higher-value oxygenates (CO, CH
3OH, HCHO, CH
3CH
2OH, etc.). The choice of oxidant is crucial for the reactivity and selectivity of PPOM, and molecular oxygen, being inexpensive and environmentally friendly, is often used as the oxidant in the PPOM process. The coupling of O
2 reduction and CH
4 oxidation is thermodynamically favorable, and the reactive oxygen species (ROS) formed from O
2 reduction facilitate CH
4 activation and product formation. Therefore, understanding the role of O
2 in the PPOM process is vital. Research in this area can be divided into gas-phase and liquid-phase systems.
2.1. Gas-Phase Systems
In gas-phase systems, methane can be photo-oxidized by O
2 to produce oxygenates such as CH
3OH, HCHO, and CO. In 1987, Brazdil’s research team first achieved the photocatalytic conversion of CH
4 to CH
3OH on CuMoO
4 [
33]. Under visible light irradiation and at 100 °C, using O
2 as the oxidant, a CH
3OH yield of 6 μmol·h
−1 was achieved. The doping of Cu
2+ extended the catalyst’s visible light activity and prolonged the lifespan of O
−. Additionally, Shuben Li’s research team achieved CH
4 photo-oxidation to CH
3OH at temperatures below 350 K and atmospheric pressure using Mo-doped porous TiO
2 catalysts pre-adsorbed with water [
34]. In 2019, Xiaoyong Wu’s research team reported a g-C
3N
4-modified Cs
0.33WO
3 photocatalyst (g-C
3N
4@Cs
0.33WO
3) that selectively photo-oxidized low-concentration CH
4 (1000 ppm) to CH
3OH at room temperature, with a yield of 4.38 μmol·g
−1·h
−1 [
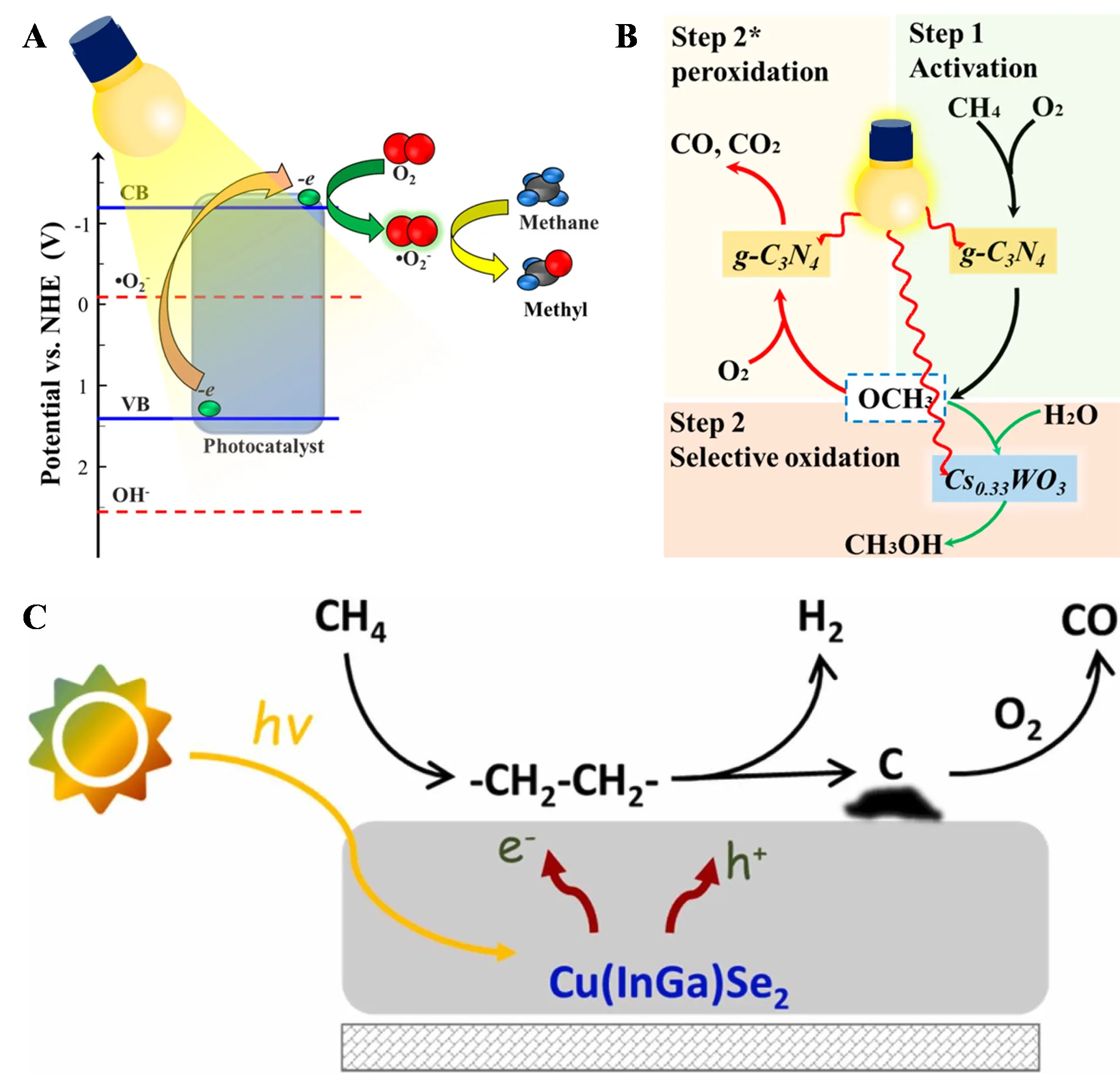
35]. As shown in A, O
2 was activated to ·O
2−, which oxidized CH
4 on the g-C
3N
4 surface to methoxy radicals. B illustrates two methoxy radical reaction pathways: in the selective oxidation pathway, photogenerated electrons from Cs
0.33WO
3 rapidly transfer to g-C
3N
4, preventing overoxidation of methoxy radicals, while the other pathway produces a small amount of CO
x, possibly due to overoxidation of CH
4 on g-C
3N
4 not bound to Cs
0.33WO
3. However, in gas-phase systems, product desorption is crucial, and the desorption of CH
3OH requires relatively high temperatures, which can lead to overoxidation. Therefore, producing liquid oxygenates (CH
3OH and HCHO) in gas-phase PPOM systems is challenging.
In contrast, gas-phase systems are more suitable for selective CO production. In 1988, Grätzel’s research team achieved the photocatalytic conversion of CH
4 to CO at room temperature and atmospheric pressure using TiO
2-supported molybdenum oxide (TiO
2/MoO
3) [
36]. Buxing Han’s research team developed an Ag/AgCl@SiO
2 photocatalyst that selectively photo-oxidized CH
4 to CO, with a CO yield of 2.3 μmol·h
−1 and a selectivity of 73% [
37]. Mechanistic studies revealed that singlet
1O
2 generated in situ from O
2 could activate methane to form the key intermediate COOH
∗, which further dehydrates to form CO. Notably, this catalyst effectively photo-oxidized CH
4 to CO using sunlight in outdoor tests, demonstrating its practical application potential.
In the above works, the hydrogen products were H
2O. In contrast, converting CH
4 to hydrogen, especially syngas (CO and H
2), is more valuable and meaningful. In 2019, Miyauchi’s research team proposed a PPOM scheme by loading Pd nanoparticles onto ultra-wide bandgap (UWBG) strontium tantalate (Sr
2Ta
2O
7) [
38]. Compared to dark conditions, light irradiation reduced the starting temperature for syngas formation to below 423 K, and under external temperatures of 623 K and UV irradiation, CO and H
2 production rates reached 46.8 mmol·g
−1·min
−1 and 54.9 mmol·g
−1·min
−1, respectively. The photothermal carriers generated by interband excitation of Pd nanoparticles drove the photocatalytic reaction, with separated hot electrons and holes promoting the activation of O
2 and CH
4, respectively, while the thermal relaxation of carriers increased the catalyst surface temperature, further facilitating the reaction. The research team also incorporated a series of noble metals (Rh, Pd, Ru, and Pt) into MCM-41 molecular sieves, with Rh/MCM performing best, achieving efficient syngas production (CO selectivity ~50%) in a flow system at temperatures as low as 423 K, with a CH
4 quantum yield of 1.8% (λ ≥ 250 nm) [
39]. Similarly, the hot electrons and holes generated by interband excitation of metal nanoparticles directly activated adsorbed O
2 and CH
4, and the photothermal effect from carrier relaxation further promoted the reaction. Recently, Ordomsky’s research team used conventional Cu(In,Ga)Se
2 (CIGS) absorbers to selectively photo-oxidize CH
4 to CO and H
2 at room temperature [
40]. The CIGS films coated on Mo showed the best performance, with a CO yield of 2.4 mmol·g
−1 and a CO/H
2 ratio of approximately 2:1, with CO selectivity exceeding 80%. As shown in C, the reaction involves stepwise dissociation and coupling of CH
4 to form hydrocarbons, followed by dehydrogenation to form disordered carbon, and finally partial oxidation of carbon to CO. While photocatalytic partial oxidation of methane to CO and H
2 reduces reaction temperature and mitigates the explosion risk of premixed CH
4/O
2, the reaction still tends to produce more stable H
2O and CO
2, resulting in low selectivity for CO and H
2, and the CO/H
2 ratio is not 1:2, making it difficult to use directly for methanol or Fischer-Tropsch synthesis. Therefore, to apply photocatalytic partial oxidation of methane to syngas in industrial production, it is necessary to improve the selectivity and ratio of CO and H
2 and further reduce the reaction temperature.
. (<b>A</b>) Proposed O<sub>2</sub> activation mechanism and (<b>B</b>) Schematic illustration for PPOM over g-C<sub>3</sub>N<sub>4</sub>@Cs<sub>0.33</sub>WO<sub>3</sub> [
35]. (<b>C</b>) Schematic diagram of CH<sub>4</sub> partial oxidation to syngas over CIGS [
40]. Note: Step 2 in B represents selective oxidation and Step 2 * represents peroxidation.
Compared to gas-phase systems, using H
2O as a solvent in liquid-phase systems offers unique advantages. Firstly, H
2O can promote catalyst dispersion. Secondly, the presence of H
2O facilitates the desorption of liquid oxygenates from the catalyst surface, significantly inhibiting the overoxidation of liquid products and thus improving reaction selectivity. Additionally, the introduction of H
2O makes O
2 activation into reactive oxygen species (ROS) easier, while H
2O itself can also serve as a source of ROS. These ROS can promote the activation of CH
4 and the formation of products, although confirming the sources of ROS adds to the complexity of research. Based on the different ROS generated from O
2 activation, current research can be categorized into several types.
2.2.1. O
2 Activation to ·OOH
In 2019, JinHua Ye’s research team first reported the use of O
2 as an oxidant to convert CH
4 into oxygenates in a liquid-phase system [
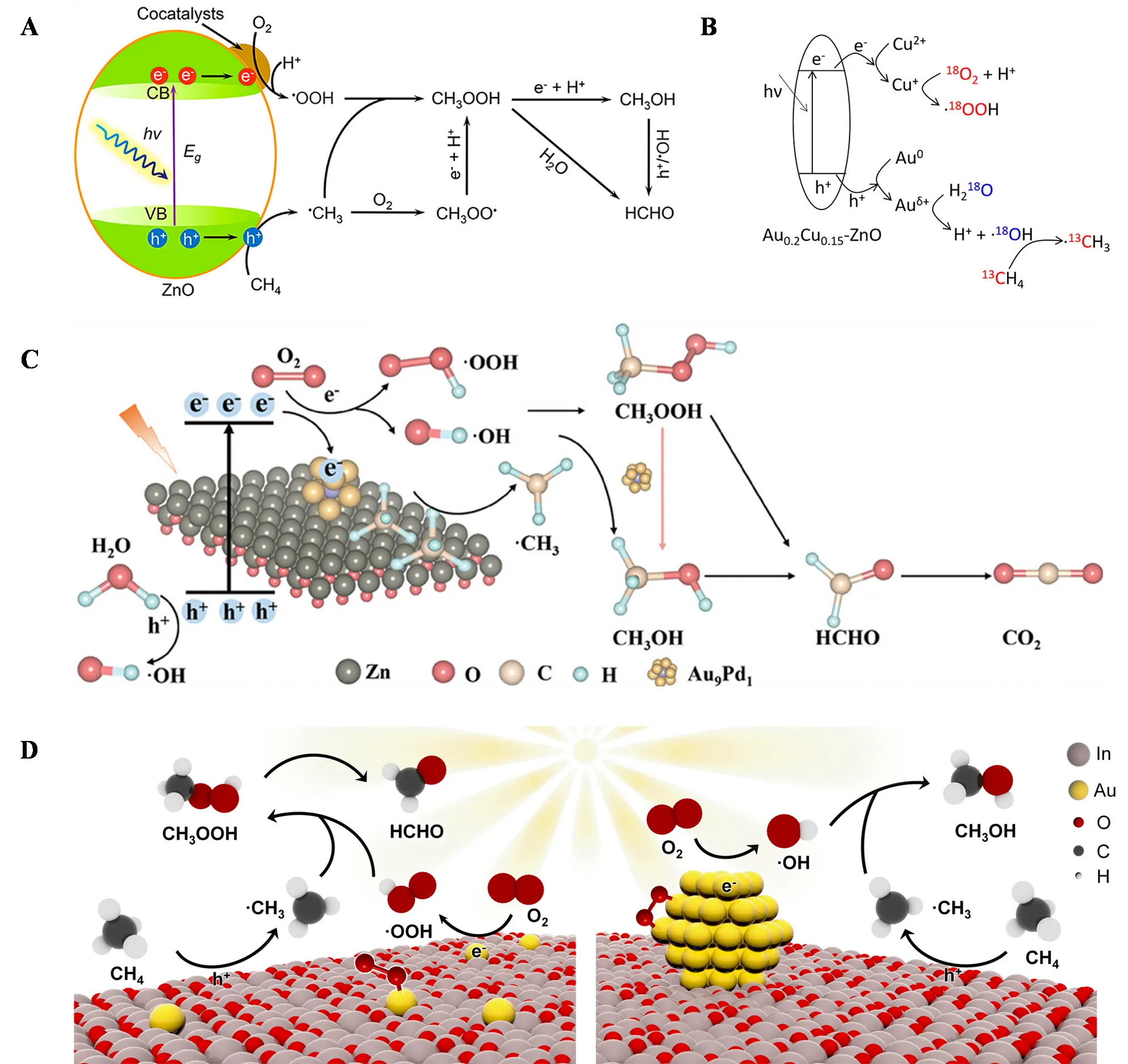
41]. Under ambient conditions and light irradiation, different cocatalysts (Pt, Pd, Au, Ag) loaded on ZnO achieved efficient production of liquid oxygenates (CH
3OH and HCHO), with Au−ZnO showing the best performance, achieving a yield of 125 μmol·h
−1 and a selectivity over 95%. Under light excitation, photogenerated holes and electrons on ZnO were separated, with CH
4 oxidized to ·CH
3 by photogenerated holes, while O
2, assisted by protonation in water, was reduced to ·OOH by photogenerated electrons on the cocatalyst. Subsequently, ·CH
3 and ·OOH combined to form the initial product CH
3OOH, which was further reduced to CH
3OH. HCHO could be produced by the photooxidation of CH
3OH by photogenerated holes or ·OH, or directly by the decomposition of CH
3OOH (A). Isotope labeling experiments further confirmed that the O in CH
3OH originated from CH
3OOH, rather than from the coupling of ·CH
3 and ·OH generated from the photooxidation of water. Subsequently, the research team developed a dual-cocatalyst modified titanium dioxide photocatalyst (Au−CoO
x/TiO
2), achieving a primary product (CH
3OH and HCHO) yield of 2540 μmol·g
−1·h
−1 and a selectivity of 95% under ambient conditions [
42]. Mechanistic studies indicated that the excellent activity and selectivity stemmed from the synergistic effect of Au nanoparticles and CoO
x. Upon illumination, Au nanoparticles facilitated the separation of photogenerated carriers and the reduction of O
2, while ·OH generated from water oxidation could over-oxidize CH
3OH to HCHO and CO
2. CoO
x modulated the oxidative capacity of the photocatalyst, inhibiting the formation of highly oxidative ·OH, thus improving selectivity.
Junwang Tang’s research team reported an Au−Cu alloy-modified ZnO (Au
0.2Cu
0.15−ZnO) achieving a primary product (CH
3OH, CH
3OOH, and HCHO) yield of 11,225 μmol·g
−1·h
−1, with nearly 100% selectivity and an apparent quantum efficiency of 14.1% at 365 nm [
43]. As shown in B, Cu acted as the electron acceptor, reducing O
2 to ·OOH, while Au accepted photogenerated holes to oxidize H
2O to ·OH, synergistically promoting charge separation and methane conversion. Both ·OH and photogenerated holes could oxidize methane to ·CH
3, and in ROS quenching experiments, the introduction of salicylic acid as a sacrificial agent for ·OH nearly halted methane conversion, proving that ·OH was the primary active species for methane activation. They also developed Pd−def−In
2O
3 [
44], Pd−def−TiO
2 [
45], Pd−def−WO
3 [
46], and Cu−def−WO
3 [
47] photocatalysts with similar PPOM mechanisms. Under light excitation, semiconductor supports generated photogenerated electron-hole pairs, with O
2 reduced to ·OOH radicals by photogenerated electrons and H
2O oxidized to ·OH by holes, further activating methane. These studies provide guidance for the rational design of future catalysts.
. (<b>A</b>) Schematic diagram of photocatalytic CH<sub>4</sub> oxidation over cocatalyst/ZnO [
41]. (<b>B</b>) Schematic illustration of photocatalytic methane conversion over Au<sub>0.2</sub>Cu<sub>0.15</sub>−ZnO [
43]. (<b>C</b>) Proposed photocatalytic mechanism for the selective oxidation of CH<sub>4</sub> over Au<sub>9</sub>Pd<sub>1</sub>/ZnO with O<sub>2</sub> [
48]. (
D) Proposed mechanism for PPOM on Au<sub>1</sub>/In<sub>2</sub>O<sub>3</sub> (left) and Au<sub>NPs</sub>/In<sub>2</sub>O<sub>3</sub> (right) [
49].
Jun Wang’s research team reported AuPd nanoparticle-loaded defect ZnO nanosheets (Au
9Pd
1/ZnO) for efficient photocatalytic methane oxidation to oxygenates under ambient conditions, achieving a maximum liquid oxygenate yield of 152.2 mM·g
−1·h
−1 with a selectivity of 86.7% and an apparent quantum efficiency of 16.5% at 380 nm [
48]. The excellent photocatalytic performance was attributed to the synergistic effect between the defect ZnO substrate and the AuPd cocatalyst. The former promoted CH
4 adsorption, while the latter enhanced light absorption, charge separation, and O
2 activation to ROS. As illustrated in C, under illumination, electrons were excited from the valence band to the conduction band of ZnO, then transferred to Au
9Pd
1 nanoparticles, reducing O
2 to ·OOH and ·OH. Photogenerated holes remaining in the ZnO valence band could oxidize CH
4 to ·CH
3 and H
2O to ·OH, although this process was less efficient. Thus, ROS generated from O
2 reduction played a dominant role in activating CH
4 adsorbed on ZnO to ·CH
3. ·CH
3 could then interact with ·OOH and ·OH to form CH
3OOH and CH
3OH. In the presence of Au
9Pd
1 nanoparticles, CH
3OOH could be partially converted to CH
3OH via a two-electron reduction process, with further oxidation by holes or ·OH leading to HCHO and CO
2 formation.
Zhiyong Tang’s research team achieved selective generation of ·OOH and ·OH by adjusting the band structure and active site size of Au/In
2O
3, enabling efficient and selective production of HCHO and CH
3OH [
49]. After three hours of photocatalytic CH
4 oxidation at room temperature, the HCHO yield on Au single-atom-loaded In
2O
3 (Au
1/In
2O
3) reached 6.09 mmol·g
−1 with a selectivity of 97.62%, while Au nanoparticle-loaded In
2O
3 (AuNPs/In
2O
3) achieved a CH
3OH yield of 5.95 mmol·g
−1 with a selectivity of 89.42%. D summarizes the entire process of selective photocatalytic oxidation of CH
4 on Au/In
2O
3. For Au
1/In
2O
3, its valence band potential is more negative than the H
2O/·OH oxidation potential, preventing ·OH generation during the reaction. Instead, photogenerated holes oxidize CH
4 to ·CH
3, while photogenerated electrons transferred to Au reduce adsorbed O
2, with end-on adsorbed O
2 favoring reduction to ·OOH. ·OOH subsequently combines with ·CH
3 to form CH
3OOH, which decomposes to form HCHO. On the surface of Au nanoparticles, side-on adsorbed O
2 is more easily reduced to ·OH, which combines with ·CH
3 to form CH
3OH. This demonstrates that rational design of photocatalysts can precisely control the types of radicals formed.
2.2.2. O
2 Activation to ·OH
In liquid-phase PPOM systems, not only can H
2O be oxidized to produce ·OH, but O
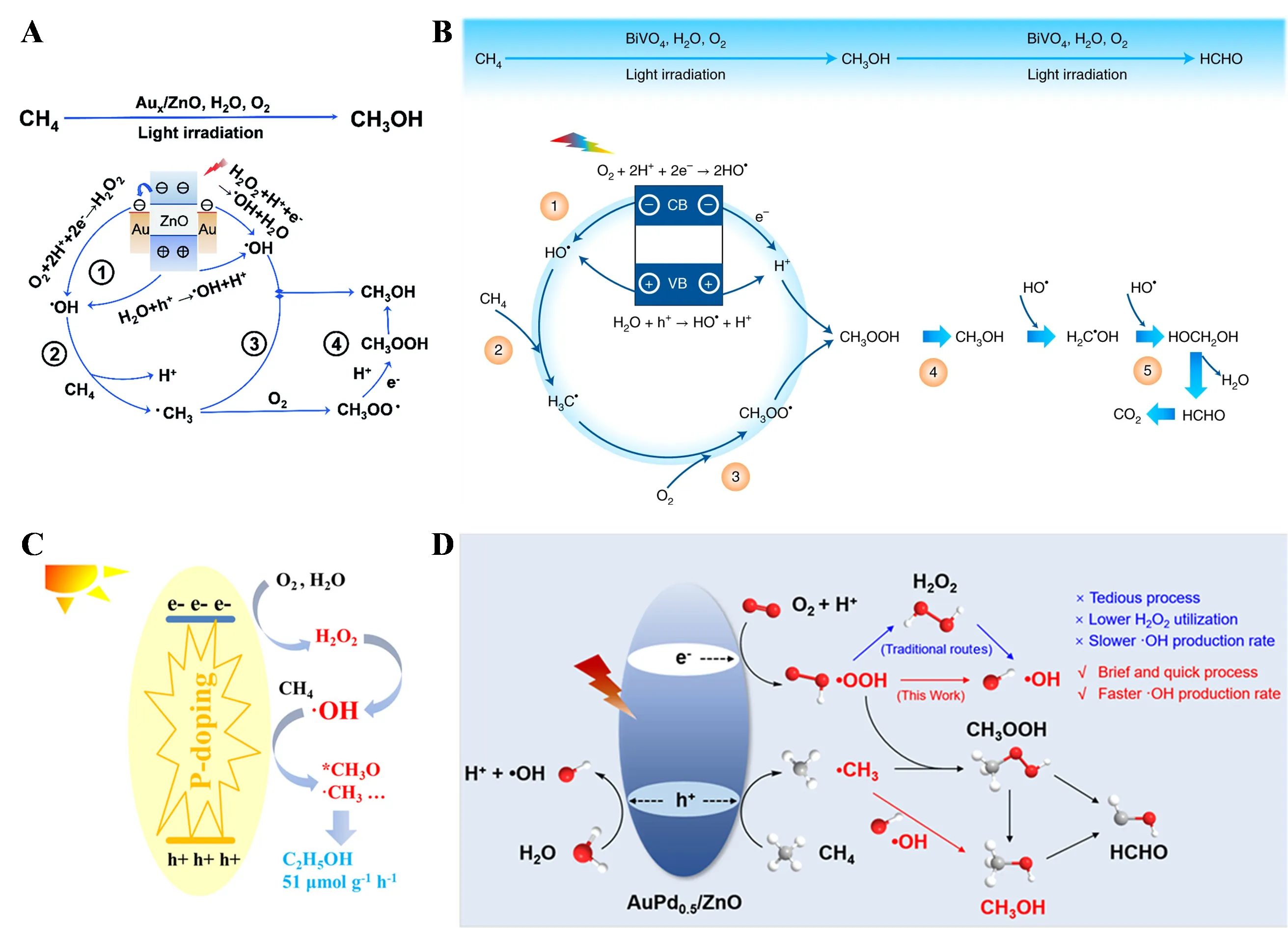
2 can also be activated to ·OH, which participates in the reaction. The ·OH radicals promote methane activation and product formation and participate in the oxidation of intermediates. However, an excess of ·OH can lead to overoxidation of products, so controlling ·OH formation is crucial for improving reaction activity and selectivity. In 2020, Zhiyong Tang’s research team designed Au nanoparticle-modified ZnO photocatalysts that, for the first time, activated O
2 to ·OH in a liquid-phase PPOM system [
50]. Under ambient conditions and light irradiation, the Au/ZnO catalyst exhibited excellent performance, with a CH
3OH yield of 1371 μmol·g
−1 and a selectivity of 99.1%. Isotope experiments with
18O demonstrated that the O atoms in the product CH
3OH originated from both O
2 and H
2O, not just O
2. As shown in A, Au nanoparticles act as electron conductors, effectively extracting electrons from the conduction band of ZnO and injecting them into O
2, producing ·OH through the O
2→H
2O
2→·OH pathway. Simultaneously, holes remaining in the valence band of ZnO oxidize H
2O to ·OH. Subsequently, CH
4 is activated by the generated ·OH to form ·CH
3, and finally, ·OH and ·CH
3 directly combine to produce CH
3OH.
They also developed quantum-sized BiVO
4 (q−BiVO
4) capable of oxidizing methane to liquid oxygenates under visible light irradiation [
51]. It was found that the selectivity for CH
3OH and HCHO could be regulated by altering the amounts of O
2 and H
2O in the reaction system, reaction time, and the wavelength and intensity of light irradiation. Shorter wavelengths and longer reaction times enhanced the oxidation ability towards CH
4, favoring HCHO formation under prolonged UV irradiation. Conversely, visible light irradiation and the introduction of large amounts of water to increase dissolved CH
4 content inhibited overoxidation, thereby increasing CH
3OH selectivity. In this system, O
2 is activated to ·OH, which activates CH
4 to form ·CH
3. ·CH
3 then combines with O
2, protons, and an electron to form CH
3OOH, which decomposes to produce CH
3OH, with further oxidation yielding HCHO (B).
JinHua Ye’s research team reported P-doped g-C
3N
4 (CNP) achieving a CH
3CH
2OH yield of 51 μmol·g
−1·h
−1 at 25 °C and 1 atm [
52]. As shown in C, P doping enhanced the process of O
2 activation through H
2O
2 (O
2→H
2O
2→·OH) to generate ·OH, which activated CH
4 to form ·CH
3. ·CH
3 further formed CH
3CH
2OH along with small amounts of HCOOH and CO
2. Wenting Wu’s research team constructed a sulfone-modified conjugated organic polymer that achieved photocatalytic conversion of CH
4 to CH
3OH and HCOOH under ambient light irradiation [
53]. Mechanistic studies showed that light irradiation induced homolysis of S=O bonds on the catalyst, generating ·O and ·S. ·O could adsorb and activate CH
4, while ·S provided electrons to
1O
2, generating H
2O
2, which then decomposed into ·OH, further oxidizing CH
4. Recently, the team also reported an Au−Pd alloy-modified ZnO photocatalyst (Au−Pd
0.5/ZnO) for CH
4 conversion to CH
3OH, achieving a CH
3OH yield and selectivity of 81.0 μmol·h
−1 and 88.2%, respectively [
54]. Unlike the traditional O
2→H
2O
2→·OH pathway, they proposed a strategy for efficiently generating ·OH directly from O
2→·OOH→·OH, improving CH
3OH yield and selectivity. As shown in D, under light irradiation, photogenerated holes oxidize CH
4 to ·CH
3, while O
2 adsorbed on the Au−Pd alloy is reduced to ·OOH by photogenerated electrons. The Au−Pd alloy facilitates O
2 adsorption and the cleavage of the O−O bond in ·OOH, quickly and directly converting ·OOH to ·OH. Finally, ·CH
3 combines with ·OH to form CH
3OH. This work not only provides a new strategy for efficiently generating ·OH directly but also offers guidance for the precise design of composite photocatalysts for PPOM reactions.
. (<b>A</b>) Schematic illustration of photocatalytic CH<sub>4</sub> conversion on Au/ZnO [
50]. (<b>B</b>) Proposed mechanism of photocatalytic CH<sub>4</sub> oxidation over q−BiVO
4<sub></sub> [
51]. (<b>C</b>) Schematic diagram of photocatalytic methane conversion over CNP [
52]. (<b>D</b>) Proposed mechanism of photocatalytic CH<sub>4</sub> oxidation on AuPd<sub>0.5</sub>/ZnO [
54].
2.2.3. O
2 Activation to ·O
2−
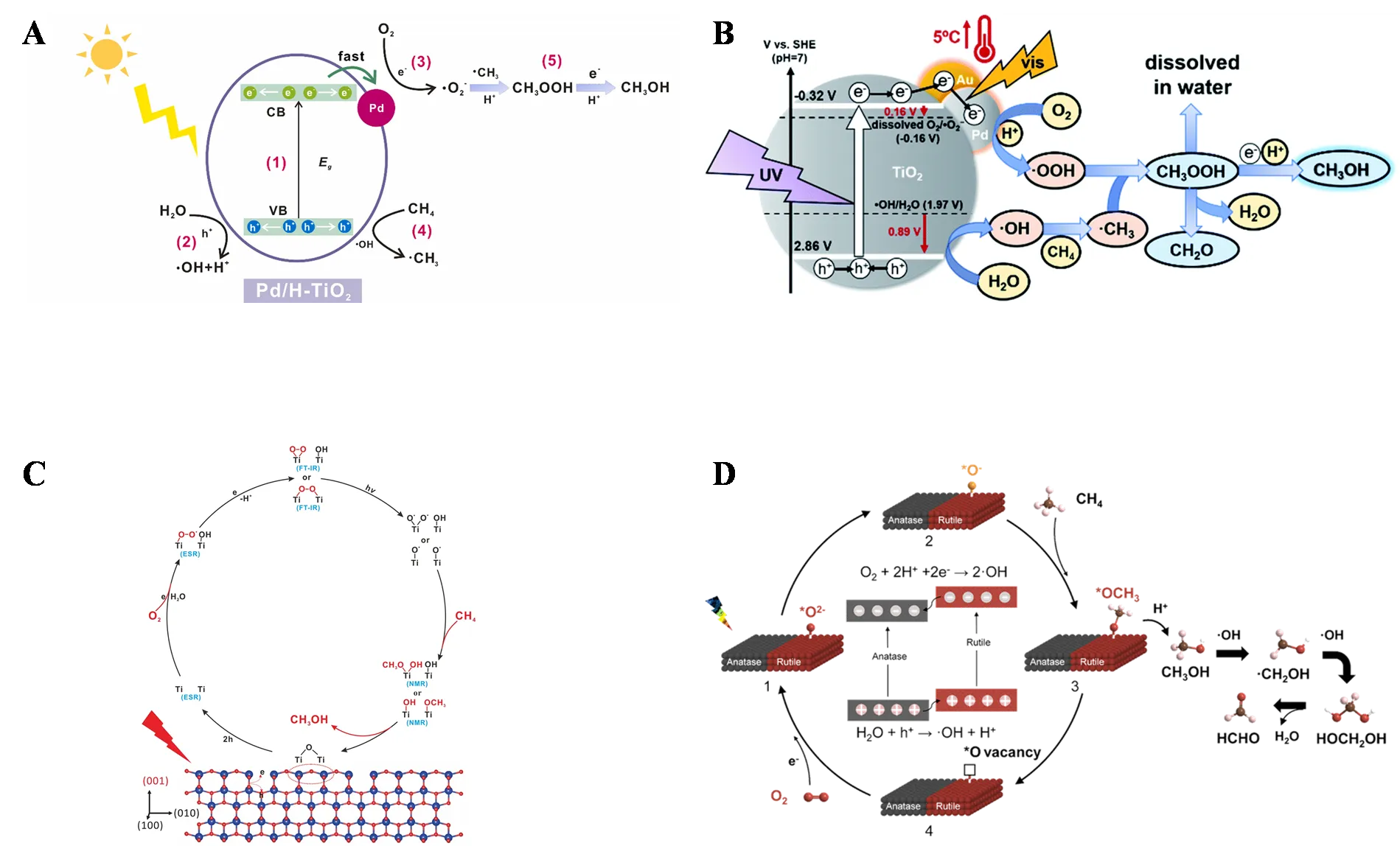
O
2 can also be activated to ·O
2−. Qin Kuang’s research team developed a hollow porous Pd/H−TiO
2 photocatalyst, where the unique hollow structure and strong metal-support interaction synergistically promoted the photocatalytic conversion of CH
4 to CH
3OH [
55]. As shown in A, ·OH and ·O
2− are the primary active species involved in the photocatalytic process. Here, ·OH primarily activates methane to ·CH
3, while ·O
2− further converts ·CH
3 to CH
3OH.
Liangshu Zhong’s research team reported a W-doped TiO
2 photocatalyst [
56]. The W doping modified the electronic and band structure of TiO
2, achieving a yield of liquid oxygenates of 12.2 mmol·g
−1 with a selectivity of 99.4%. Subsequently, they developed a Cu and W co-doped TiO
2 (Cu−W−TiO
2) photocatalyst, achieving an oxygenate yield of 34.5 mmol·g
−1 with a selectivity of 97.1% [
57]. Under light irradiation, O
2 was reduced to ·O
2− by photogenerated electrons or electrons captured in the W
6+/W
5+ cycle, while CH
4 was activated to ·CH
3 by Cu
+. The ·O
2− combined with H
+ to form ·OOH, which reacted with ·CH
3 to form CH
3OOH, which was further reduced to CH
3OH. The synergistic effect of hole and electron capture processes on Cu−W−TiO
2 reduced the recombination of photogenerated carriers, promoting methane activation and efficient conversion. They also developed a SrWO
4/TiO
2 heterojunction catalyst [
58]. The formation of the heterostructure facilitated the separation and transfer of photogenerated carriers, achieving an oxygenate yield of 13,365 μmol·g
−1 and a selectivity of 98.7%. Yunhang Hu’s research team reported an Au−Pd/TiO
2 photocatalyst, achieving a CH
3OH yield of 12.6 mmol·g
−1·h
−1 in the presence of O
2 and H
2O [
59]. As shown in B, photogenerated electrons reduced O
2 to ·O
2−, which was then converted to ·OOH. Meanwhile, H
2O was oxidized to ·OH by photogenerated holes, activating CH
4 to ·CH
3. ·CH
3 coupled with ·OOH to form CH
3OOH, which was further reduced to CH
3OH. TiO
2 absorbed UV light, generating electrons and holes, while Au−Pd nanoparticles not only facilitated the transfer of photogenerated electrons but also absorbed visible light, increasing the catalyst temperature. The increased temperature enhanced the process of H
2O oxidation to ·OH and drove the reduction of O
2 to ·O
2− and the conversion of CH
3OOH to CH
3OH. The synergistic effect of Au−Pd nanoparticles and TiO
2 facilitated the efficient conversion of CH
4 to CH
3OH.
Li Niu’s research team prepared Au nanoparticle-modified cubic WO
3 (c−WO
3) for the selective photo-oxidation of CH
4 to HCHO [
60].
18O
2 isotope tests indicated that the O in HCHO originated from lattice oxygen on the exposed (002), (020), and (200) planes of c−WO
3, and the surface-consumed lattice oxygen could be regenerated by the reduction of O
2. Under light irradiation, W
5+ and O
− were generated on the c−WO
3 surface. O
− cleaved the C—H bond, activating CH
4 to −OCH
3, which was further dehydrogenated by adjacent terminal O
− to form HOCH
2OH, and finally dehydrated to form HCHO. Au nanoparticles captured photogenerated electrons, further facilitating the formation of O
−. JinHua Ye’s research team achieved a CH
3OH yield of 4.8 mmol·g
−1·h
−1 with a selectivity of about 80% using Ag-modified TiO
2 with a dominant (001) facet to inhibit PPOM overoxidation [
61]. As shown in C, on the TiO
2(001) surface, photogenerated holes oxidized surface oxygen to form oxygen vacancies. O
2 reduced by photogenerated electrons formed ·O
2−, which were stabilized by oxygen vacancies, forming surface superoxides (Ti−O
2·). These could capture photogenerated electrons, forming surface peroxides (Ti−OO−Ti and Ti−(OO)), which dissociated into Ti−O· pairs. These pairs could directly activate CH
4 to form CH
3OH, effectively avoiding the formation of ·CH
3 and ·OH, thus inhibiting overoxidation.
Zhiyong Tang’s research team used a “pause-flow” reactor with a TiO
2 dual-phase catalyst (anatase 90% and rutile 10%) (anatase/rutile) for the highly selective conversion of CH
4 to HCHO, achieving an HCHO yield of 8.09 mmol·g
−1·h
−1 with a selectivity of 97.4% [
62]. Under light irradiation, carriers were generated inside A/R−TiO
2, with O
2 activated to ·O
2− on R−TiO
2. The ·O
2− was further activated to O
− species, activating CH
4 to
∗CH
3O.
∗CH
3O was converted to CH
3OH, desorbed with the assistance of H
2O, and finally oxidized to HCHO by ·OH. The O
− species originated from TiO
2 lattice oxygen, with O
2 filling the oxygen vacancies in TiO
2 (D). This work guides the rational design of catalysts and reactors for industrial photocatalytic conversion of low-carbon feedstocks and demonstrates the feasibility of large-scale formaldehyde production.
In the PPOM process, controlling the selectivity for single oxygenate products remains a significant challenge. O
2 activation can produce various ROS, each with different oxidation capabilities and mechanisms for activating CH
4. Future research should focus on the rational design of photocatalysts to precisely control the type of ROS generated, thereby controlling intermediate formation to enhance reaction efficiency and selectivity. Additionally, current photocatalytic methane partial oxidation reactions primarily occur in closed systems. While some progress has been made, the limitations of closed systems restrict large-scale production of oxygenates. Therefore, future research should focus on developing reaction systems and designing reactors to achieve efficient methane conversion on a larger scale, paving the way for industrial photocatalytic methane conversion.
. (<b>A</b>) Schematic illustration for the oxidation of methane to methanol over Pd/H−TiO<sub>2</sub> [
55]. (<b>B</b>) Schematic diagram of photocatalytic methane conversion over Au–Pd/TiO<sub>2</sub> [
59]. (<b>C</b>) Proposed mechanism for CH<sub>4</sub> oxidation by O<sub>2</sub> on the (001) facets of TiO<sub>2</sub> [
61]. (<b>D</b>) Proposed mechanism of photocatalytic methane oxidation on A/R−TiO<sub>2</sub> [
62].
3. Photocatalytic Oxidative Coupling of Methane (POCM)
The oxidative coupling of methane (OCM) refers to the conversion of methane into C
2+ hydrocarbons in the presence of an oxidant (typically O
2) [
63]. Since Keller and Bhasin first reported the OCM production of valuable chemicals like ethylene and ethane in 1982 [
64], the OCM reaction has garnered increasing attention. However, while the introduction of O
2 lowers the Gibbs free energy of the reaction, it still requires relatively high reaction temperatures and inevitably generates overoxidized products such as CO and CO
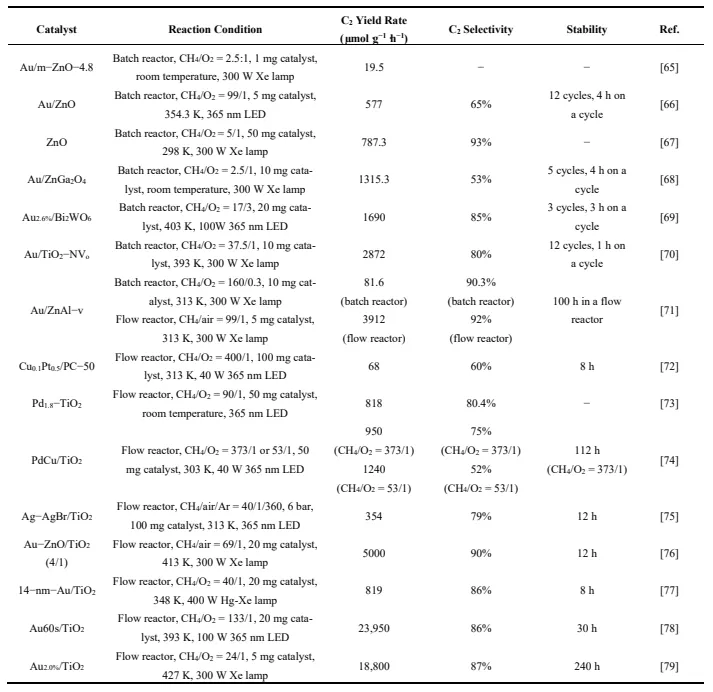
2. Therefore, photocatalytic oxidative coupling of methane (POCM) under mild conditions has become a new research hotspot. Researchers have deeply studied catalyst design, reactor optimization, and reaction mechanisms, achieving some significant results ().
. Representative works on photocatalytic OCM reaction with O2 as the oxidant.
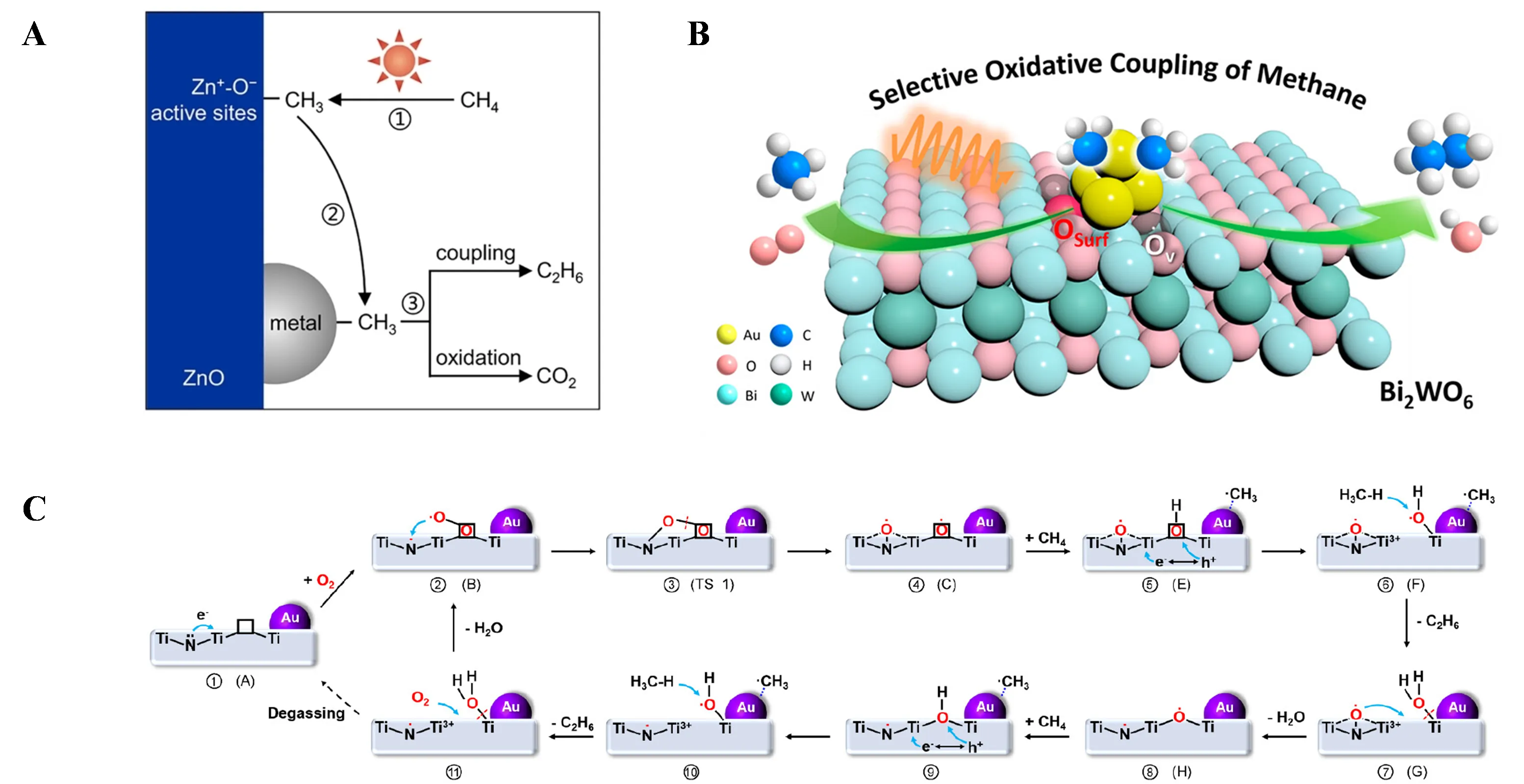
In 2018, Jinlin Long’s research team achieved room-temperature photocatalytic coupling of methane to ethane using Au nanoparticles supported on porous ZnO nanosheets (Au/m−ZnO−4.8) [
65]. The plasmonic field formed at the Au/ZnO interface effectively induced charge separation in photo-excited ZnO, initiating methane activation at the Zn sites. Mechanistic studies revealed that the rate-determining step for methane coupling was the reduction of protons by hot electrons induced by Au plasmonics. The introduction of O
2 generated reactive oxygen species (ROS), accelerating proton consumption and increasing the ethane production rate from 11.0 µmol·g
−1·h
−1 to 19.5 µmol·g
−1·h
−1. Tierui Zhang’s research team systematically elucidated the photocatalytic OCM mechanism by designing a series of transition metal (Au, Ag, Pd, Cu, Ni, Ru, and Pt)-supported ZnO nanoparticles (M/ZnO) [
66]. They found that whether
∗CH
3 underwent C—C coupling or deep dehydrogenation on the metal surface was closely related to the d−σ interaction between
∗CH
3 and the metal. Au/ZnO achieved the highest C
2−C
4 yield of 683 µmol·g
−1·h
−1 (selectivity 83%) due to strong d-σ hybridization between
∗CH
3 and Au, which reduced the Au—C—H bond angle, decreasing the spatial hindrance for
∗CH
3 coupling along the C—C pathway. As shown in A, photoexcited Zn
+−O
− active sites on ZnO efficiently activated methane to
∗CH
3 and adsorbed H
2O, followed by selective coupling of
∗CH
3 on the metal surface to form ethane or overoxidation to CO
2. Meanwhile, O
2 was reduced by photogenerated electrons, following the pathway O
2→O
2−→O
22−→2O
−→2O
2−, replenishing the Ov created by H
2O desorption on ZnO. Scholten’s research team synthesized ZnO nanostructures with photocatalytic OCM activity using the hydrolysis of imidazole zinc chloride ionic liquids (ILs). The size and shape (irregular particles, nanorods) of the ZnO nanostructures depended on the synthesis conditions [
67]. When the ionic liquid to ZnCl
2 ratio was equimolar, the resulting irregular ZnO particles had the highest C
2H
6 yield (787.3 µmol·g
−1·h
−1) and a selectivity of about 93%. This result provides a new approach to designing efficient POCM catalysts under room temperature and non-metal conditions.
In addition to using classic ZnO semiconductors as POCM supports, researchers have explored other high-activity supports, achieving some progress. In 2023, Li Li’s research team used Au nanoparticle-modified ZnGa
2O
4 nanosheets (Au/ZnGa
2O
4) for the photocatalytic oxidative coupling of methane, achieving an ethane yield of 1315.3 µmol·g
−1·h
−1 and a selectivity of 53% [
68]. The results showed that the reaction activity significantly increased compared to pure ZnGa
2O
4 due to Au nanoparticles promoting O
2 adsorption and activation, producing O
2−. O
2− was further reduced by photogenerated electrons to O
−, which cracked CH
4 to ·CH
3. Au nanoparticles effectively stabilized ·CH
3 and prevented its overoxidation to CO
2. Similarly, Dunwei Wang’s research team achieved an ethane yield of 1690 µmol·g
−1·h
−1 and a selectivity of 85% by constructing an Au
2.6%/Bi
2WO
6 model to regulate ROS [
69]. The study found that OCM performance was highly sensitive to the properties of the photocatalyst, which should facilitate the release of surface lattice oxygen, forming oxygen vacancies more easily. Au cocatalysts enhanced lattice oxygen activity, promoting Ov formation. Bi
2WO
6 has tunable surface oxygen, and under light irradiation, lattice oxygen was excited, activating the C—H bond in CH
4. The resulting ·CH
3 migrated to Au sites, where another CH
4 molecule was activated, producing a desorbable H
2O molecule and leaving an Ov. O
2 filled the Ov (B). Throughout the process, O
2− was the active species, so ·CH
3 was more likely to couple at Au sites to form C2H6 rather than over-oxidize to CO
2.
Zizhong Zhang’s research team achieved high activity and selectivity for photocatalytic OCM by constructing dual-active sites (N and oxygen vacancies) on TiO
2 nanosheets, regulating O
2 activation pathways [
70]. Due to the different O
2 activation sites on TiO
2, the alkane yield on TiO
2 nanosheets with N and Ov dual-active sites (Au/TiO
2−NV
o) increased from 1600 µmol·g
−1·h
−1 to 3200 µmol·g
−1·h
−1, and the selectivity improved from 61% to 93% compared to regular Au/TiO
2 nanosheets. For Au/TiO
2, O
2 easily captured photogenerated electrons on TiO
2, forming O
2−, which tended to react with ·CH
3 intermediates to form
−OOCH
3, which further decomposed into H
2O and CO
2, leading to CH
4 overoxidation. For Au/TiO
2−NVo, as shown in C, O
2 adsorbed on oxygen vacancies was reduced to O
2−, and under the action of excited N atoms, the O−O bond of O
2− cleaved, forming milder O
·− active species. O
·− could cleave the C—H bond in CH
4 to form ·CH
3, and hydroxyl radicals formed by the photolysis of Ti−O bonds activated the second CH
4. ·CH
3 coupled on Au NPs to form C
2H
6. After O atoms refilled the oxygen vacancies, the process continued with another two CH
4 molecules, inhibiting CH
4 overoxidation and enhancing OCM activity and selectivity. Unlike traditional oxide catalysts for POCM reactions, Yufei Song’s research team achieved an ethane yield of 81.6 µmol·g
−1·h
−1 and a selectivity of 90.3% by constructing Au-loaded ZnAl layered double hydroxide with oxygen vacancies (Au/ZnAl-v) [
71]. In a batch system, the catalytic activity was stable for 100 h in a flow system, with a CH
4 conversion rate of 8.5 mmol·g
−1·h
−1 and a C
2H
6 selectivity of 92%. The study found that introducing Ov into ZnAl-LDH significantly promoted the efficiency and selectivity of photocatalytic OCM. O
2 first adsorbed and activated on Ov, then the activated O
2 cleaved the C—H bond in CH
4. The resulting ·CH
3 migrated to Au sites, coupling to form ethane. This report provides a new approach to designing efficient, selective, and stable photocatalytic materials for OCM using non-traditional oxide supports.
. (<b>A</b>) Schematic diagram of the proposed reaction mechanism for photocatalytic OCM on metal/ZnO [
66]. <b>(B</b>) Schematic description of Au-modified Bi<sub>2</sub>WO<sub>6</sub> nanosheets for POCM [
69]. (<b>C</b>) Proposed reaction mechanism of the POCM on Au/TiO<sub>2</sub>−NV<sub>o</sub> [
70].
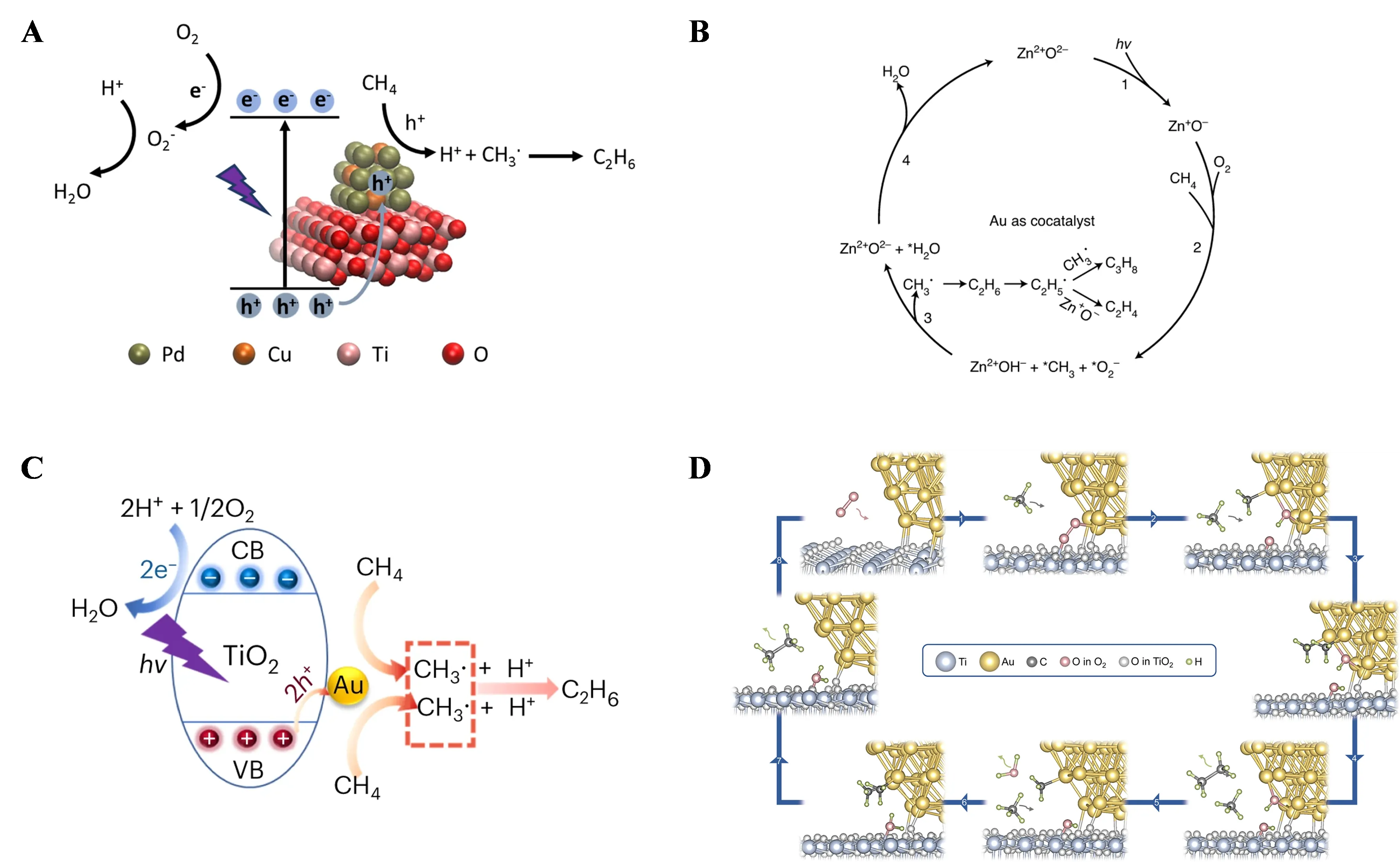
In batch systems, although gas-catalyst contact is sufficient, prolonged residence time inevitably leads to overoxidation of CH
4, resulting in lower selectivity. Additionally, scaling up batch reactors to industrial production is challenging. Therefore, developing flow reaction systems is essential for achieving efficient and selective photocatalytic OCM.
In 2020, Junwang Tang’s research team first reported photocatalytic OCM in a flow reaction system [
72]. They introduced Pt nanoparticles and CuO
x clusters onto TiO
2 (PC-50) (Cu
0.1Pt
0.5/PC-50), achieving a C
2 hydrocarbon yield of 68 µmol·g
−1·h
−1 and a selectivity of 60% under ambient conditions. The synergistic effect of Pt nanoparticles and CuO
x clusters increased the C
2 hydrocarbon yield by 3.5 times compared to PC-50 and more than twice the combined activity of Pt/PC-50 and Cu/PC-50. Under light irradiation, electrons were excited from the valence band to the conduction band of TiO
2 and migrated to Pt, while photogenerated holes transferred to CuO
x clusters. This process delayed carrier recombination and lowered the oxidation potential of photogenerated holes, preventing overoxidation of CH
4. CH
4 was activated by photogenerated holes on CuO
x clusters, forming ·CH
3 and H
+, with ·CH
3 coupling to form C
2H
6. Pt nanoparticles reduced O
2 and combined H
+ to remove it as H
2O.
Similarly, the research team used Pd nanoparticle-modified anatase TiO
2 (Pd
1.8−TiO
2) to achieve a C
2H
6 yield of 818 µmol·g
−1·h
−1 under mild conditions, which was 13 times that of pure TiO
2, with a selectivity of 80.4% [
73]. Pd nanoparticles acted as photogenerated hole acceptors, participating in CH
4 activation and ·CH
3 coupling, effectively inhibiting photogenerated carrier recombination, thereby significantly improving catalyst performance. They further reported a highly efficient and stable PdCu nanoalloy-modified TiO
2 (PdCu/TiO
2) photocatalyst for OCM in a mild flow system, achieving a C
2 hydrocarbon yield of 1240 µmol·g
−1·h
−1 and the photocatalyst exhibits the turnover frequency and turnover number of 116 h
−1 and 12,642 with respect to PdCu. [
74]. As shown in A, under light irradiation, photogenerated electrons from TiO
2 reduced O
2 to superoxide radicals, while photogenerated holes transferred to the PdCu alloy to activate adsorbed CH
4, generating ·CH
3 and H
+. ·CH
3 further coupled to form C
2H
6, with H
+ consumed by superoxide radicals to form H
2O. Introducing Pd nanoparticles into TiO
2 led to effective charge transfer, weakening the C—H bond in CH
4 and facilitating its activation, producing more ·CH
3. Cu reduced the adsorption energy of the target product C
2H
6, preventing catalyst coking. Thus, the synergistic effect of PdCu nanoalloy achieved efficient, selective, and stable photocatalytic OCM. Additionally, they designed an Ag−AgBr/TiO
2 ternary catalyst and studied the effect of reaction pressure on photocatalytic OCM performance in a pressurized flow reactor [
75]. When the reaction pressure increased from 1 bar to 6 bar, the C
2H
6 yield increased from 18.2 µmol·g
−1·h
−1 to 354 µmol·g
−1·h
−1, with a C
2+ selectivity of 79%. Higher reaction pressure enhanced the mass transfer efficiency of reactants and products, improving reaction efficiency and selectivity. Ag nanoparticles, as electron acceptors, facilitated charge transfer, while the AgBr and TiO
2 heterostructure reduced oxidation capability, preventing overoxidation. Thus, designing high-performance catalysts and rational reaction systems is crucial for photocatalytic OCM.
Au NPs, as a cocatalyst, not only promote charge separation but also stabilize ·CH
3, preventing CH
4 overoxidation to CO
2, thereby improving photocatalytic OCM activity and selectivity. Moreover, the electromagnetic decay of localized surface plasmon resonance (LSPR) on Au nanoparticles can generate hot carriers. Hot carrier relaxation can induce a photothermal effect and the hot carrier separation not only promotes O
2 reduction and CH
4 activation, but also prolongs carrier lifetime. Therefore, using Au NPs as cocatalysts in photocatalytic OCM is favored by researchers. In 2021, JinHua Ye’s research team efficiently and selectively coupled CH
4 to C
2H
6 using Au NP-loaded ZnO/TiO
2 hybrids (Au−ZnO/TiO
2(4/1)), achieving a C
2H
6 yield of over 5000 µmol·g
−1·h
−1 with 90% selectivity [
76]. The study found that modifying ZnO with TiO
2 and Au formed a ZnO/TiO
2−Au heterojunction, enhancing photocatalytic activity while maintaining ZnO’s mild C—H bond overoxidation capability in CH
4. Introducing Au cocatalysts promoted O
2 adsorption and activation, facilitating the desorption of
∗CH
3 as ·CH
3 in the gas phase, thus promoting C
2H
6 formation and inhibiting overoxidation to CO
2. As shown in B, under light irradiation, the photocatalyst generated photogenerated electrons and holes, with the heterojunction allowing rapid electron transfer to Au NPs, reducing O
2 to
∗O
2−, while photogenerated holes transferred to ZnO, aiding CH
4 activation to produce
∗CH
3.
∗CH
3 desorbed as ·CH
3 on Au and coupled to form C
2H
6. C
2H
6 further reacted with holes to form ·C
2H
5, which coupled with ·CH
3 to form C
3H
8 or further oxidized to C
2H
4, with H
2O as a byproduct. Unlike Au, Pt, with stronger O
2 reduction capability, tended to form
∗OCH
3, ultimately oxidizing to CO
2. A lower O
2/CH
4 ratio meant less collision between
∗O and
∗CH
3, inhibiting CH
4 overoxidation. Therefore, the O
2/CH
4 ratio in feed gas significantly controls product selectivity.
Subsequently, Khodakov’s research team studied the role of 6–60 nm plasmonic Au NPs supported on TiO
2 in methane oxidative coupling. Under optimized conditions with 14 nm Au NPs (14−nm−Au/TiO
2), an ethane yield of 819 µmol·g
−1·h
−1 and a selectivity of 86% were achieved [
77]. They found that the size (6–60 nm) and amount (>0.5 wt%) of Au NPs did not significantly affect methane coupling. Under UV excitation, TiO
2 generated oxygen vacancies, activating methane at these sites, while Au NPs activated O
2 and facilitated charge separation. Using a rapid sputtering method, Junwang Tang’s research team synthesized highly efficient Au60s/TiO
2 catalysts on glass fiber filters, achieving a C
2 hydrocarbon yield of 23,950 µmol·g
−1·h
−1 with 86% selectivity, the highest reported efficiency to date [
78]. Introducing Au NPs as cocatalysts extended the lifetime of TiO
2 photoelectrons by 66 times, forming more superoxide radicals, promoting the photocatalytic methane conversion cycle. As catalytic centers and photogenerated hole acceptors, Au NPs facilitated methane adsorption and increased photogenerated hole numbers, favoring selective C—H bond cleavage and C—C bond coupling. Under light irradiation, TiO
2 generated photogenerated electrons and holes, with photogenerated holes transferring to Au NPs while long-lived photogenerated electrons on TiO
2 reduced oxygen. Au efficiently adsorbed and activated CH
4, generating ·CH
3 and H
+, with ·CH
3 coupling to form C
2H
6 and H
+ combining with superoxide radicals to form H
2O (C).
Recently, Yujie Xiong’s research team loaded Au NPs onto TiO
2 nanosheets (Au
2.0%/TiO
2), achieving a C
2+ hydrocarbon yield of 19,280 µmol·g
−1·h
−1 with 90% selectivity in a custom 3D-printed multi-point injection flow reactor, maintaining stability for over 240 h [
79]. Unlike pure TiO
2, where O
2− induced CH
4 overoxidation, the presence of Au NPs stabilized
∗CH
3 intermediates and constructed an Au−TiO
2 interface, regulating O
2 activation to produce mild O
22− species, avoiding
∗CH
3 overoxidation. The localized electric field induced by Au NPs’ LSPR promoted the polarization and dissociation of C—H bonds in CH
4. As shown in D, under light irradiation, photogenerated electrons in the TiO
2 conduction band were captured by Au NPs, achieving O
2 activation at the Au−TiO
2 interface, generating mild O
22− species. These mild O
22− species dissociated adsorbed CH
4 on Au NPs to produce
∗CH
3. Another CH
4 underwent a similar process to generate
∗CH
3, which coupled to form C
2H
6. The two formed
∗OH radicals continued to abstract hydrogen atoms from two other CH
4 molecules, generating a second C
2H
6 and two H
2O molecules, regenerating O
2 activation sites, completing the photocatalytic OCM cycle. This work guides the synergistic design of reactors and photocatalysts to simultaneously regulate mass transfer and reactant activation for high-performance flow systems.
In summary, future research should focus on reaction system and photocatalyst design. For reaction systems, flow systems have improved reaction efficiency and target product selectivity, but issues such as low mass transfer efficiency and product separation remain. Designing more efficient reactors to control the catalytic conversion process is necessary. Additionally, the CH
4/O
2 ratio, flow rate, and reaction pressure significantly impact photocatalytic OCM performance. Therefore, ensuring safety requires finer control of reaction conditions. For photocatalyst design, rational catalysts should generate mild active oxygen species to promote methane activation while stabilizing methyl intermediates to facilitate C—C bond coupling. Currently, most cocatalysts are Au or other precious metals, and the main product is ethane, which has lower commercial value. Considering economic feasibility, developing non-precious metal catalysts and novel catalysts to regulate CH
4 activation and carbon intermediate conversion to produce higher-value chemicals such as ethylene and propane is essential.
. (<b>A</b>) Schematic diagram of photoexcitation and reaction over PdCu/TiO<sub>2</sub> [
74]. (<b>B</b>) Schematic illustration of the reaction pathways for photocatalytic OCM over Au−ZnO/TiO<sub>2</sub>(4/1) [
76]. (<b>C</b>) Proposed POCM process over Au60s/TiO<sub>2</sub> [
78]. (<b>D</b>) Schematic illustration of the proposed mechanism for photocatalytic OCM on Au<sub>2.0%</sub>/TiO<sub>2</sub> [
79].
4. Photochemical Looping
Chemical looping involves decomposing a reaction into multiple sub-reactions, which are carried out in isolated spaces to achieve in situ separation of products [
80,
81,
82,
83]. In methane aerobic conversion reactions, the use of oxygen can lead to deep oxidation of methane and poses an explosion risk, significantly limiting the industrial application of this reaction. Applying chemical looping techniques to methane conversion, dividing the reaction into two half-reactions, can greatly improve the selectivity and safety of the reaction. This technology typically uses metal oxides as oxygen carriers. In the methane oxidation reaction, lattice oxygen is used instead of traditional O
2. Subsequently, the reduced oxygen carrier reacts with air to replenish the lattice oxygen, completing one reaction loop. Yong Lu’s research team achieved methane oxidative coupling at lower temperatures through chemical looping to activate O
2 [
84]. They developed a TiO
2-doped Mn
2O
3−Na
2WO
4/SiO
2 catalyst system, generating MnTiO
3 in the reaction flow, triggering the MnTiO
3↔Mn
2O
3 chemical loop to activate O
2, achieving a 22% CH
4 conversion rate and 62% C
2−C
3 selectivity at 650 °C. Liang-Shih Fan’s research team embedded Fe
2O
3 nanoparticles in a mesoporous SiO
2 carrier (Fe
2O
3@SBA-15), significantly suppressing CO
2 production in methane partial oxidation, achieving nearly 100% CO selectivity in a cyclic redox system at 750–935 °C [
85]. Theoretical calculations indicated that low-coordination Fe atoms favored CH
4 adsorption and activation, while low-coordination lattice oxygen atoms significantly promoted Fe−O bond cleavage and CO formation, thereby enhancing CO selectivity.
Based on the characteristics of chemical looping, researchers have applied it to photocatalytic systems, yielding valuable results. In 2012, Jiesheng Chen’s research team first reported a Ga
3+-modified ETS-10 molecular sieve material that utilized its oxygen centers and metal center active sites for strong activation of methane C—H bonds, achieving efficient coupling of methane to ethane at room temperature [
86]. Under light irradiation, photogenerated electrons reduced Ti
4+ to Ti
3+, while photogenerated holes oxidized surface hydroxyl groups to hydroxyl radicals, thus activating the C—H bonds of methane. As the reaction proceeded, Ti−OH groups were gradually consumed, leading to catalyst deactivation, which could be restored to photocatalytic activity by simple heat treatment in humid air.
In 2020, Khodakov’s research team proposed a photochemical looping strategy using highly dispersed silver ions in a silver-phosphotungstic acid-titanium dioxide nanocomposite (Ag−HPW/TiO
2) to achieve high selectivity and nearly quantitative ethane production [
87]. As Ag
+ was continuously reduced to metallic Ag, the catalyst’s color changed from light gray to deep black, gradually deactivating the catalyst. To continuously synthesize ethane, the catalyst was regenerated by exposing it to air under light for 7 h. As shown in A, under light irradiation, the photocatalyst generated photogenerated electrons and holes. The electrons reduced Ag
+ to metallic Ag, while the holes oxidized CH
4 to ·CH
3, which coupled to form C
2H
6. Exposing the catalyst to air under light regenerated Ag to its oxidized state, forming an Ag
+↔Ag
0 redox loop. This strategy separated the CH
4 oxidation step from the O
2 reduction step, effectively inhibiting methane overoxidation.
Similarly, Yongfu Sun’s research team achieved efficient CH
4→C
2H
6 photocatalytic conversion using Au/ZnO porous nanosheets with dual active species of Au
δ− and O
− [
88]. During photoexcitation, lattice oxygen in ZnO was easily oxidized to O
− active species. The generated Au
δ− and O
− could polarize the inert C—H bond and stabilize the resulting active
∗CH
3 intermediate, thus avoiding overoxidation. The consumed lattice oxygen was replenished through the Mars-van Krevelen mechanism. Ye Wang’s research team constructed a solar-driven Fe
3+/Fe
2+ redox loop, combining CH
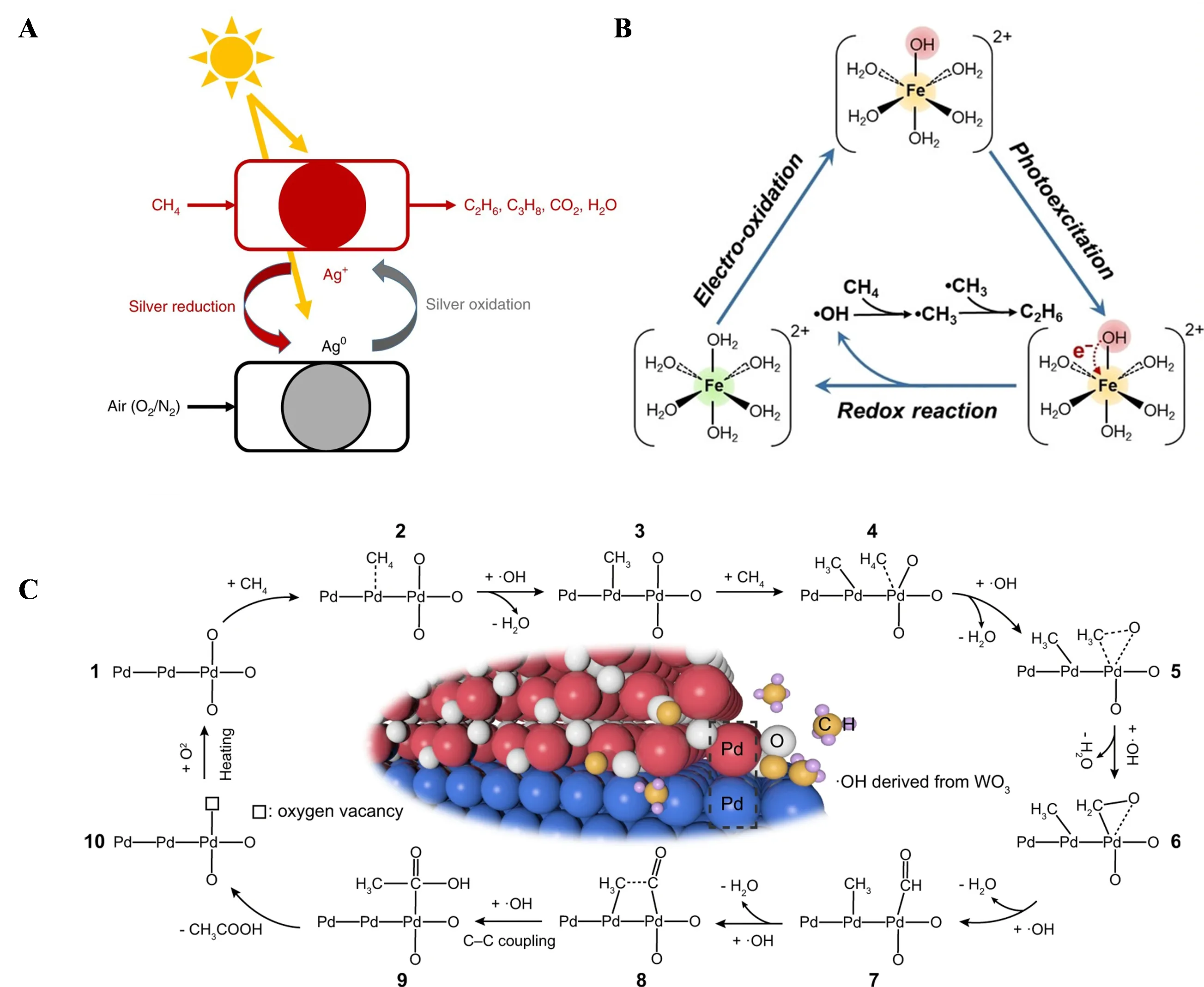
4 photochemical coupling with electrochemical H
2 production. This system achieved high selectivity for CH
4 coupling to C
2H
6 while reducing the potential for electrochemical hydrogen production [
89]. Fe
3+ hydrolyzed to form [Fe(H
2O)
5OH]
2+, which absorbed UV light, transferring electrons from the OH to Fe
3+, resulting in free
∗OH and Fe
2+.
∗OH oxidized CH
4 to
∗CH
3, which coupled to form C
2H
6. Fe
2+ was oxidized back to Fe
3+ at the anode, replacing the traditional oxygen evolution reaction (OER), and H
2 was produced at the cathode, completing the redox loop (B).
Yujie Xiong’s research team proposed a methoxy- and ethoxy-intermediate-mediated pathway, directly photocatalytically converting CH
4 to C
2H
4 under mild conditions using ZnO−AuPd
2.7% [
90]. Zn sites on ZnO served as the adsorption and activation sites for CH
4, activating it to
∗CH
3. The resulting
∗CH
3 preferred to combine with ZnO lattice oxygen to form methoxy intermediates. Assisted by Pd, methoxy could dehydrogenate to −CH
2O, which then reacted with another activated CH
4 to form ethoxy intermediates. Ethoxy further dehydrogenated to form C
2H
4, avoiding the overoxidation of CH
4 to CO
x. After 8 h, 3.68% of ZnO total lattice oxygen was consumed, reducing catalytic activity. The formed oxygen vacancies were easily replenished by washing with water. They also designed Pd−Zn modified WO
3 nanosheets (Pd
5/Zn
0.35−WO
3) to achieve efficient conversion of CH
4 to C
2H
4 [
91]. Zn sites promoted the adsorption and activation of CH
4, forming methyl and methoxy intermediates with the help of lattice oxygen. Pd sites facilitated methoxy dehydrogenation to methylene radicals, forming C
2H
4 and inhibiting overoxidation. The consumed lattice oxygen could be replenished by photochemical looping with air exposure. Additionally, they reported a Pd single-atom modified TiO
2 photocatalyst (Pd
1/TiO
2) for methane coupling to ethane [
92]. For TiO
2, the significant contribution of O atoms to the valence band made lattice oxygen directly involved in methane activation. The activated
∗CH
3 was difficult to desorb from O sites, leading to overoxidation. In Pd1/TiO
2, the Pd−O4 unit contributed most to the surface TiO
2 valence band maximum (VBM), accumulating photogenerated holes and facilitating CH
4 dissociation on Pd, inhibiting overoxidation with lattice oxygen. However, this process still required lattice oxygen consumption, leading to performance degradation after 6 h. The consumed lattice oxygen could be replenished by heating in air. Recently, they loaded Au NPs on Nb3O7(OH) with abundant surface lattice hydroxyl groups, achieving efficient photocatalytic coupling of CH
4 to C
2H
6 under mild conditions [
93]. Lattice hydroxyls on Nb3O7(OH) facilitated CH
4 activation, forming key methoxy intermediates. The consumed lattice oxygen could be replenished by photochemical looping, either by washing with water or air exposure.
Photochemical looping strategies can also be applied to the partial oxidation of methane to produce oxygenates. In 2019, Khodakov’s research team highly dispersed zinc on phosphotungstic acid/titanium dioxide (Zn−HPW/TiO
2), selectively photocatalytically oxidizing methane to carbon monoxide under ambient conditions [
94]. Under UV excitation, ZnO formed Zn
+−O
− pairs that adsorbed and activated methane to form Zn-methyl species. Surface methyl zinc reacted with zinc carbonate (formed from ZnO and gas-phase CO
2) to form methyl zinc carbonate, which decomposed to produce CO. After 12 h, the formation rates of CO and CO
2 slowed due to insufficient oxygen content and gradual reduction of Zn
2+ to Zn
0. The Zn
2+ was regenerated and lattice oxygen replenished through the Mars-van Krevelen mechanism by irradiation in air. Photochemical looping can also occur in gas-solid-liquid systems. Yujie Xiong’s research team constructed a PdO/Pd−WO
3 heterojunction nanocomposite with CH
4 activation and C—C coupling active sites, directly converting CH
4 to CH
3COOH without introducing additional carbon sources [
95]. As shown in C, CH
4 adsorbed on Pd sites was activated by ·OH, with
∗CH
3 gradually converting to Pd−CO intermediates under the assistance of O atoms in PdO and ·OH dehydrogenation.
∗CO and
∗CH
3 coupled to form Pd−COCH
3 intermediates, which further hydrolyzed to CH
3COOH. The formation of CH
3COOH consumed lattice oxygen in PdO, leading to performance decay after 3 h. The consumed lattice oxygen could be replenished by heating in air.
Currently, photochemical looping strategies for methane conversion are applied only in closed systems and not connected to air systems. Most applications are limited to batch systems. Future research should focus on reactor design, especially flow reaction systems. Additionally, the time-consuming Mars-van Krevelen mechanism for catalyst regeneration needs to be made more efficient for industrial applications. Moreover, hydrogen products are mostly water. Adjusting the reaction pathways of active species to produce more valuable hydrogen gas would be beneficial. Combining photochemical looping strategies with methane conversion could solve existing challenges and realize the industrialization of photocatalytic methane conversion.
. (<b>A</b>) Schematic diagram of a photochemical looping process on Ag−HPW/TiO<sub>2</sub> [
87]. (<b>B</b>) Schematic diagram of the Fe<sup>3+</sup>−Fe<sup>2+</sup> cycle pathway and mechanism [
89]. (<b>C</b>) Schematic illustration for photochemical conversion of CH<sub>4</sub> to CH<sub>3</sub>COOH over PdO/Pd−WO<sub>3</sub> [
95].
5. Summary and Outlook
Directly converting methane into value-added chemicals offers a promising alternative to the energy-intensive industrial methane reforming processes. Compared to the thermodynamically unfavorable non-oxidative coupling of methane (NOCM), the introduction of oxygen not only lowers the Gibbs free energy of the reaction, making it spontaneous, but also produces various reactive oxygen species that facilitate methane activation. Photocatalysis can reduce the activation barrier for methane, activating the inert C—H bond and enabling the reaction under milder conditions. This review has summarized the latest research progress in the photocatalytic partial oxidation of methane (PPOM) and oxidative coupling of methane (POCM) using oxygen as an oxidant. It primarily covers the activation mechanisms of methane and oxygen in different reaction systems, evaluation of methane aerobic conversion efficiency, and elucidation of reaction mechanisms. Additionally, the unique advantages of photochemical cycling in methane conversion are introduced. These insights aim to deepen the understanding of methane and oxygen activation mechanisms to design more efficient catalysts and reaction systems.
Despite substantial progress in photocatalytic methane conversion, significant gaps remain before industrial application can be realized. For example, the catalytic performance obtained experimentally is far from the requirements of industrial production; the high-carbon products is difficult to be synthesized efficiently and directionally; the poor selectivity of the products caused by peroxidation makes it difficult to separate the products; the reaction system is relatively elementary and the reactor is difficult to scale up; the studies of the reaction mechanism is insufficient, especially the activation mechanism of the C—H bond as well as the monitoring of the reaction intermediates and the reactive oxygen species. To address these issues, future research should focus on the following aspects:
(1) Designing Efficient Catalysts: Currently, most photocatalysts for methane conversion are UV-responsive. Therefore, designing narrow-bandgap semiconductors or black photocatalysts that absorb the entire solar spectrum, including visible and infrared light, is essential. Photocatalysts should also possess good electron-hole separation capabilities and suitable redox potentials. They should have moderate oxidation ability for methane and strong activation capacity for oxygen to promote the formation of reactive oxygen species. Typically, precious metals are introduced as cocatalysts to promote charge separation and oxygen activation. However, for economic viability, doping, defect engineering, heterojunction construction, or developing non-precious metal catalysts to replace precious metal catalysts should be explored. Traditional inorganic semiconductor materials often limit photocatalysts, so developing new photocatalytic materials, including covalent organic frameworks and inorganic-organic or metal-organic frameworks, may show excellent performance in methane conversion. Moreover, hot-carrier mediated photocatalysis can be induced by the Landau damping of surface plasmon resonance (SPR) in plasmonic metals or direct interband electron transitions in other noble metals [
96,
97]. Hot carriers can not only undergo relaxation to increase the catalyst surface temperature, but also directly participate in the photochemical reaction process, which greatly promotes the reaction efficiency. Therefore, by establishing a plasmonic or non-plasmonic hot carrier-based photocatalytic system can make isolating support materials exhibit photocatalytic activity, which undoubtedly enriches the photocatalyst system and has significant meaning for finding new photocatalyst materials.
(2) Hot carriers’ effect: The role of metal nanoparticles is usually considered to be the promotion of light absorption, charge separation, activation of reactants, and stabilization of reaction intermediates. However, metal nanoparticles can simultaneously utilize the photo and thermal energy to promote reaction. The hot carriers’ effect induced by metal nanoparticles should not be ignored for photocatalytic conversion of methane. More attention should be paid to how the hot carriers’ effect promotes the reduction of O
2 and the activation of CH
4 to improve the efficiency and selectivity of the reaction. The electromagnetic decay of localized surface plasmon resonance (LSPR) on plasmonic nanoparticles such as Au, Ag and Cu or interband electron transition from the d-band to the s-band in non-plasmonic nanoparticles such as Pt, Pd, Rh, and Ru can produce hot carriers, which are different from those carriers generated by traditional semiconductor bandgap excitation [
98,
99]. In general, bandgap excitation and hot carrier generation compete with each other. Therefore, it is possible to promote the generation of hot carriers by choosing a suitable metal as well as by regulating the supports. Hot carrier relaxation can induce a photothermal effect to increase the catalyst surface temperature. Moreover, hot carriers can directly interact with gas molecules adsorbed on the surface of metal nanoparticles and thus participate in photochemical processes. Meanwhile, the process of hot carrier charge separation not only prolongs the carrier lifetime, but also promotes the reduction of O
2 and the activation of CH
4, which further improves the efficiency and selectivity of the reaction. Therefore, it is necessary to fully understand and utilize the hot carriers’ effect of metal nanoparticles.
(3) Expanding to C
2+ Products: The products of photocatalytic methane aerobic conversion are mostly C
1 oxygenates or C
2H
6, which have low commercial value and require further conversion for industrial significance. Reports on C
2 oxygenates, C
2H
4, and C
3H
8 are rare and yields are low. Methane activation energy is usually higher than that of oxidation products, leading to overoxidation of products. Oxidation and coupling reactions compete, making it challenging to form C
2+ products. This necessitates deeper mechanistic studies to reveal possible reaction pathways and precise control of reaction sites and intermediates to promote C—C bond coupling.
(4) Developing Efficient Reaction Systems: The reaction system is critical for reaction activity and product selectivity. Although flow systems have advantages over batch systems in terms of gas-solid or gas-solid-liquid mass transfer efficiency, reaction stability, and selectivity, the complexity of methane conversion and the costs and safety of reactions should be considered. Drawing from mature flow systems in thermal catalysis, developing novel photocatalytic flow reactors is essential. Currently reported photocatalytic methane conversion flow systems mainly apply to gas-solid two-phase reactions. Gas-liquid-solid three-phase systems, such as those in methane partial oxidation, largely remain in batch reactors. Developing new reactors, such as membrane reactors, can further enhance methane reaction efficiency and target product selectivity. Reaction temperature, pressure, flow rate, CH
4/O
2 ratio, light source, and reaction time all affect yield and product selectivity. For example, an appropriate CH
4/O
2 ratio can prevent overoxidation in methane coupling, and pressurized reactors can increase the solubility of CH
4 and O
2 in H
2O. Additionally, due to the low selectivity of products in methane aerobic conversion, the cost of subsequent product separation is high. Rationally designing porous metal-organic frameworks can replace energy-intensive distillation for separation and purification.
(5) Utilizing Advanced In Situ Characterization: Research on the reaction mechanisms of photocatalytic methane aerobic conversion heavily relies on molecular and atomic-level characterization techniques, especially in situ and operando characterization. Since oxygen activation can produce various reactive oxygen species, in situ EPR can detect these species to understand the mechanism of O
2 activation. Isotope labeling can determine the source of liquid oxygen. Advanced techniques like in situ IR spectroscopy, in situ XPS, and X-ray absorption spectroscopy can characterize the chemical states of reaction intermediates and active sites. Time-resolved spectroscopy can provide precise information on methane activation and conversion processes. Theoretical calculations can simulate reaction processes at the molecular level, exploring the reaction mechanism of methane conversion and determining the energies of different reaction steps, thus revealing the correct reaction pathway to guide the development of high-performance photocatalytic methane conversion systems.
In summary, abundant and inexpensive methane not only holds the potential to replace petroleum and other fossil fuels but can also serve as a raw material for synthesizing value-added chemicals. Due to methane’s stability, activating and converting methane molecules is highly challenging. Activating methane’s inert C—H bond using solar energy is strategically significant for energy development and sunlight utilization. Although there is still a gap between photocatalytic methane conversion and industrial application, achieving this will bring immense benefits to society.
Author Contributions
Conceptualization, Y.K. and L.L.; Methodology, Y.K.; Writing—Original Draft Preparation, Y.K. and C.Y.; Writing—Review & Editing, X.M. and L.L.; Project Administration, X.M., Y.C. and L.L.; Funding Acquisition, L.L.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Funding
This work was financially supported by the Natural Science Foundation of Jilin Province (grant nos. 20240101177JC and 20240302098GX), the National Natural Science Foundation (NSFC) of China (grant no. 22379050), the National Key Research and Development Program of China (2023YFA1506303), and the Fundamental Research Funds for the Central Universities.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
1.
Schwach P, Pan X, Bao X. Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects.
Chem. Rev. 2017,
117, 8497–8520.
[Google Scholar]
-
2.
Gunsalus NJ, Koppaka A, Park SH, Bischof SM, Hashiguchi BG, Periana RA. Homogeneous Functionalization of Methane.
Chem. Rev. 2017,
117, 8521–8573.
[Google Scholar]
-
3.
Ma Z, Chen Y, Gao C, Xiong Y. A Minireview on the Role of Cocatalysts in Semiconductor-Based Photocatalytic CH
4 Conversion.
Energy Fuels 2022,
36, 11428–11442.
[Google Scholar]
-
4.
Schwarz H. Chemistry with Methane: Concepts Rather than Recipes.
Angew. Chem. Int. Ed. 2011,
50, 10096–10115.
[Google Scholar]
-
5.
Kang Y, Tian P, Li J, Wang H, Feng K. Methane mitigation potentials and related costs of China’s coal mines. Fundam. Res. 2023. https://doi.org/10.1016/j.fmre.2023.09.012.
-
6.
Jiang Y, Li S, Fan X, Tang Z. Recent advances on aerobic photocatalytic methane conversion under mild conditions.
Nano Res. 2023,
16, 12558–12571.
[Google Scholar]
-
7.
Baltrusaitis J, Jansen I, Schuttlefield Christus JD. Renewable energy based catalytic CH
4 conversion to fuels.
Catal. Sci. Technol. 2014,
4, 2397–2411.
[Google Scholar]
-
8.
Zhan Q, Kong Y, Wang X, Li L. Photocatalytic non-oxidative conversion of methane.
Chem. Commun. 2024,
60, 2732–2743.
[Google Scholar]
-
9.
Song H, Meng X, Wang Z-j, Liu H, Ye J. Solar-Energy-Mediated Methane Conversion.
Joule 2019,
3, 1606–1636.
[Google Scholar]
-
10.
Wang P, Zhang X, Shi R, Zhao J, Yuan Z, Zhang T. Light-Driven Hydrogen Production from Steam Methane Reforming via Bimetallic PdNi Catalysts Derived from Layered Double Hydroxide Nanosheets.
Energy Fuels 2022,
36, 11627–11635.
[Google Scholar]
-
11.
Mu X, Li L. Photo-Induced Activation of Methane at Room Temperature.
Acta Phys. -Chim. Sin. 2019,
35, 968–976.
[Google Scholar]
-
12.
Angeli SD, Monteleone G, Giaconia A, Lemonidou AA. State-of-the-art catalysts for CH
4 steam reforming at low temperature.
Int. J. Hydrogen Energy 2014,
39, 1979–1997.
[Google Scholar]
-
13.
Khodakov AY, Chu W, Fongarland P. Advances in the Development of Novel Cobalt Fischer-Tropsch Catalysts for Synthesis of Long-Chain Hydrocarbons and Clean Fuels.
Chem. Rev. 2007,
107, 1692–1744.
[Google Scholar]
-
14.
Behrens M, Studt F, Kasatkin I, Kühl S, Hävecker M, Abild-Pedersen F, et al. The Active Site of Methanol Synthesis over Cu/ZnO/Al
2O
3 Industrial Catalysts.
Science 2012,
336, 893–897.
[Google Scholar]
-
15.
Ab Rahim MH, Armstrong RD, Hammond C, Dimitratos N, Freakley SJ, Forde MM, et al. Low temperature selective oxidation of methane to methanol using titania supported gold palladium copper catalysts.
Catal. Sci. Technol. 2016,
6, 3410–3418.
[Google Scholar]
-
16.
Agarwal N, Freakley SJ, McVicker RU, Althahban SM, Dimitratos N, He Q, et al. Aqueous Au−Pd colloids catalyze selective CH
4 oxidation to CH
3OH with O
2 under mild conditions.
Science 2017,
358, 223–227.
[Google Scholar]
-
17.
Deng J, Chen P, Xia S, Zheng M, Song D, Lin Y, et al. Advances in Oxidative Coupling of Methane.
Atmosphere 2023,
14, 1538.
[Google Scholar]
-
18.
Xu Y, Bao X, Lin L. Direct conversion of methane under nonoxidative conditions.
J. Catal. 2003,
216, 386–395.
[Google Scholar]
-
19.
Liu J, Yue J, Lv M, Wang F, Cui Y, Zhang Z, et al. From fundamentals to chemical engineering on oxidative coupling of methane for ethylene production: A review.
Carbon Resour. Convers. 2022,
5, 1–14.
[Google Scholar]
-
20.
Xu Y, Chen E, Tang J. Photocatalytic methane conversion to high-value chemicals.
Carbon Future 2024,
1, 9200004.
[Google Scholar]
-
21.
Zhang L, Liu L, Pan Z, Zhang R, Gao Z, Wang G, et al. Visible-light-driven non-oxidative dehydrogenation of alkanes at ambient conditions.
Nat. Energy 2022,
7, 1042–1051.
[Google Scholar]
-
22.
Tan R, Wang X, Kong Y, Ji Q, Zhan Q, Xiong Q, et al. Liberating C—H Bond Activation: Achieving 56% Quantum Efficiency in Photocatalytic Cyclohexane Dehydrogenation.
J. Am. Chem. Soc. 2024,
146, 14149–14156.
[Google Scholar]
-
23.
Li L, Mu X, Liu W, Mi Z, Li C-J. Simple and Efficient System for Combined Solar Energy Harvesting and Reversible Hydrogen Storage.
J. Am. Chem. Soc. 2015,
137, 7576–7579.
[Google Scholar]
-
24.
Li L, Li G-D, Yan C, Mu X-Y, Pan X-L, Zou X-X, et al. Efficient Sunlight-Driven Dehydrogenative Coupling of Methane to Ethane over a Zn
+-Modified Zeolite.
Angew. Chem. Int. Ed. 2011,
50, 8299–8303.
[Google Scholar]
-
25.
Li L, Fan S, Mu X, Mi Z, Li C-J. Photoinduced Conversion of Methane into Benzene over GaN Nanowires.
J. Am. Chem. Soc. 2014,
136, 7793–7796.
[Google Scholar]
-
26.
Zhang J, Shen J, Li D, Long J, Gao X, Feng W, et al. Efficiently Light-Driven Nonoxidative Coupling of Methane on Ag/NaTaO
3: A Case for Molecular-Level Understanding of the Coupling Mechanism.
ACS Catal. 2023,
13, 2094–2105.
[Google Scholar]
-
27.
Wang G, Mu X, Li J, Zhan Q, Qian Y, Mu X, et al. Light-Induced Nonoxidative Coupling of Methane Using Stable Solid Solutions.
Angew. Chem. Int. Ed. 2021,
60, 20760–20764.
[Google Scholar]
-
28.
Wang G, Mu X, Tan R, Pan Z, Li J, Zhan Q, et al. Fabrication of Stepped CeO
2 Nanoislands for Efficient Photocatalytic Methane Coupling.
ACS Catal. 2023,
13, 11666–11674.
[Google Scholar]
-
29.
Kaliaguine SL, Shelimov BN, Kazansky VB. Reactions of methane and ethane with hole centers O
−.
J. Catal. 1978,
55, 384–393.
[Google Scholar]
-
30.
Wang P, Shi R, Zhao J, Zhang T. Photodriven Methane Conversion on Transition Metal Oxide Catalyst: Recent Progress and Prospects.
Adv. Sci. 2024,
11, 2305471.
[Google Scholar]
-
31.
Yang Z, Zhang Q, Song H, Chen X, Cui J, Sun Y, et al. Partial oxidation of methane by photocatalysis.
Chin. Chem. Lett. 2024,
35, 108418.
[Google Scholar]
-
32.
Song H, Ye J. Direct photocatalytic conversion of methane to value-added chemicals.
Trends Chem. 2022,
4, 1094–1105.
[Google Scholar]
-
33.
Ward MD, Brazdil JF, Mehandru SP, Anderson AB. Methane photoactivation on copper molybdate: An experimental and theoretical study.
J. Phys. Chem. 2002,
91, 6515–6521.
[Google Scholar]
-
34.
Chen X, Li S. Photooxidation of Methane to Methanol by Molecular Oxygen on Water-preadsorbed Porous TiO
2-based Catalysts.
Chem. Lett. 2000,
29, 314–315.
[Google Scholar]
-
35.
Li Y, Li J, Zhang G, Wang K, Wu X. Selective Photocatalytic Oxidation of Low Concentration Methane over Graphitic Carbon Nitride-Decorated Tungsten Bronze Cesium.
ACS Sustain. Chem. Eng. 2019,
7, 4382–4389.
[Google Scholar]
-
36.
Thampi KR, Kiwi J, Grtzel M. Room temperature photo-activation of methane on TiO
2 supported molybdena.
Catal. Lett. 1988,
1, 109–116.
[Google Scholar]
-
37.
Zhai J, Zhou B, Wu H, Jia S, Chu M, Han S, et al. Selective photocatalytic aerobic oxidation of methane into carbon monoxide over Ag/AgCl@SiO
2.
Chem. Sci. 2022,
13, 4616–4622.
[Google Scholar]
-
38.
Jiang H, Peng X, Yamaguchi A, Ueda S, Fujita T, Abe H, et al. Photocatalytic Partial Oxidation of Methane on Palladium-Loaded Strontium Tantalate.
Sol. RRL 2019,
3, 190076.
[Google Scholar]
-
39.
Jiang H, Peng X, Yamaguchi A, Fujita T, Abe H, Miyauchi M. Synergistic photothermal and photochemical partial oxidation of methane over noble metals incorporated in mesoporous silica.
Chem. Commun. 2019,
55, 13765–13768.
[Google Scholar]
-
40.
Dong C, Hu D, Ben Tayeb K, Simon P, Addad A, Trentesaux M, et al. Photocatalytic partial oxidation of methane to carbon monoxide and hydrogen over CIGS solar cell.
Appl. Catal. B Environ. 2023,
325, 122340.
[Google Scholar]
-
41.
Song H, Meng X, Wang S, Zhou W, Wang X, Kako T, et al. Direct and Selective Photocatalytic Oxidation of CH
4 to Oxygenates with O
2 on Cocatalysts/ZnO at Room Temperature in Water.
J. Am. Chem. Soc. 2019,
141, 20507–20515.
[Google Scholar]
-
42.
Song H, Meng X, Wang S, Zhou W, Song S, Kako T, et al. Selective Photo-oxidation of Methane to Methanol with Oxygen over Dual-Cocatalyst-Modified Titanium Dioxide.
ACS Catal. 2020,
10, 14318–14326.
[Google Scholar]
-
43.
Luo L, Gong Z, Xu Y, Ma J, Liu H, Xing J, et al. Binary Au−Cu Reaction Sites Decorated ZnO for Selective Methane Oxidation to C
1 Oxygenates with Nearly 100% Selectivity at Room Temperature.
J. Am. Chem. Soc. 2021,
144, 740–750.
[Google Scholar]
-
44.
Luo L, Fu L, Liu H, Xu Y, Xing J, Chang C-R, et al. Synergy of Pd atoms and oxygen vacancies on In
2O
3 for methane conversion under visible light.
Nat. Commun. 2022,
13, 2930.
[Google Scholar]
-
45.
Gong Z, Luo L, Wang C, Tang J. Photocatalytic Methane Conversion to C
1 Oxygenates over Palladium and Oxygen Vacancies Co-Decorated TiO
2.
Sol. RRL 2022,
6, 2200335.
[Google Scholar]
-
46.
Wang K, Luo L, Wang C, Tang J. Photocatalytic methane activation by dual reaction sites co-modified WO
3.
Chin. J. Catal. 2023,
46, 103–112.
[Google Scholar]
-
47.
Luo L, Han X, Wang K, Xu Y, Xiong L, Ma J, et al. Nearly 100% selective and visible-light-driven methane conversion to formaldehyde via. single-atom Cu and W
δ+.
Nat. Commun. 2023,
14, 2690.
[Google Scholar]
-
48.
Chen F, Zhou H, Liu D, Qin X, Jing Y, Chen L, et al. Defective ZnO Nanoplates Supported AuPd Nanoparticles for Efficient Photocatalytic Methane Oxidation to Oxygenates.
Adv. Energy Mater. 2024,
14, 2303642.
[Google Scholar]
-
49.
Jiang Y, Li S, Wang S, Zhang Y, Long C, Xie J, et al. Enabling Specific Photocatalytic Methane Oxidation by Controlling Free Radical Type.
J. Am. Chem. Soc. 2023,
145, 2698–2707.
[Google Scholar]
-
50.
Zhou W, Qiu X, Jiang Y, Fan Y, Wei S, Han D, et al. Highly selective aerobic oxidation of methane to methanol over gold decorated zinc oxide via photocatalysis.
J. Mater. Chem. A 2020,
8, 13277–13284.
[Google Scholar]
-
51.
Fan Y, Zhou W, Qiu X, Li H, Jiang Y, Sun Z, et al. Selective photocatalytic oxidation of methane by quantum-sized bismuth vanadate.
Nat. Sustain. 2021,
4, 509–515.
[Google Scholar]
-
52.
Du X, Yang Z, Yang X, Zhang Q, Liu L, Ye J. Efficient Photocatalytic Conversion of Methane into Ethanol over P-Doped g-C
3N
4 under Ambient Conditions.
Energy Fuels 2022,
36, 3929–3937.
[Google Scholar]
-
53.
An B, Zhang Qh, Zheng Bs, Li M, Xi Yy, Jin X, et al. Sulfone-Decorated Conjugated Organic Polymers Activate Oxygen for Photocatalytic Methane Conversion.
Angew. Chem. Int. Ed. 2022,
61, e202204661.
[Google Scholar]
-
54.
Zhou Q, Tan X, Wang X, Zhang Q, Qi C, Yang H, et al. Selective Photocatalytic Oxidation of Methane to Methanol by Constructing a Rapid O
2 Conversion Pathway over Au–Pd/ZnO.
ACS Catal. 2024,
14, 955–964.
[Google Scholar]
-
55.
Zhang X, Wang Y, Chang K, Yang S, Liu H, Chen Q, et al. Constructing hollow porous Pd/H−TiO
2 photocatalyst for highly selective photocatalytic oxidation of methane to methanol with O
2.
Appl. Catal. B Environ. 2023,
320, 121961.
[Google Scholar]
-
56.
Huang M, Zhang S, Wu B, Yu X, Gan Y, Lin T, et al. Highly Selective Photocatalytic Aerobic Oxidation of Methane to Oxygenates with Water over W-doped TiO
2.
ChemSusChem 2022,
15, e202200548.
[Google Scholar]
-
57.
Huang M, Zhang S, Wu B, Wei Y, Yu X, Gan Y, et al. Selective Photocatalytic Oxidation of Methane to Oxygenates over Cu−W−TiO
2 with Significant Carrier Traps.
ACS Catal. 2022,
12, 9515–9525.
[Google Scholar]
-
58.
Huang M, Zhang S, Gan Y, Liu J, He Z, Lin T, et al. Effective SrWO
4/TiO
2 Heterojunction with Enhanced Carriers Separation and Transfer for Photocatalytic Methane Oxidation.
Chem. Eur. J. 2023,
29, e202204031.
[Google Scholar]
-
59.
Cai X, Fang S, Hu YH. Unprecedentedly high efficiency for photocatalytic conversion of methane to methanol over Au–Pd/TiO
2−what is the role of each component in the system?
J. Mater. Chem. A 2021,
9, 10796–10802.
[Google Scholar]
-
60.
Wei S, Zhu X, Zhang P, Fan Y, Sun Z, Zhao X, et al. Aerobic oxidation of methane to formaldehyde mediated by crystal-O over gold modified tungsten trioxide via photocatalysis.
Appl. Catal. B Environ. 2021,
283, 119661.
[Google Scholar]
-
61.
Feng N, Lin H, Song H, Yang L, Tang D, Deng F, et al. Efficient and selective photocatalytic CH
4 conversion to CH
3OH with O
2 by controlling overoxidation on TiO
2.
Nat. Commun. 2021,
12, 4652.
[Google Scholar]
-
62.
Jiang Y, Zhao W, Li S, Wang S, Fan Y, Wang F, et al. Elevating Photooxidation of Methane to Formaldehyde via TiO
2 Crystal Phase Engineering.
J. Am. Chem. Soc. 2022,
144, 15977–15987.
[Google Scholar]
-
63.
Farrell BL, Igenegbai VO, Linic S. A Viewpoint on Direct Methane Conversion to Ethane and Ethylene Using Oxidative Coupling on Solid Catalysts.
ACS Catal. 2016,
6, 4340–4346.
[Google Scholar]
-
64.
Keller GE, Bhasin MM. Synthesis of ethylene via oxidative coupling of methane: I. Determination of active catalysts.
J. Catal. 1982,
73, 9–19.
[Google Scholar]
-
65.
Meng L, Chen Z, Ma Z, He S, Hou Y, Li H-H, et al. Gold plasmon-induced photocatalytic dehydrogenative coupling of methane to ethane on polar oxide surfaces.
Energy Environ. Sci. 2018,
11, 294–298.
[Google Scholar]
-
66.
Wang P, Shi R, Zhao Y, Li Z, Zhao J, Zhao J, et al. Selective Photocatalytic Oxidative Coupling of Methane via Regulating Methyl Intermediates over Metal/ZnO Nanoparticles.
Angew. Chem. Int. Ed. 2023,
62, e202304301.
[Google Scholar]
-
67.
Souza JD, Souza VS, Scholten JD. Synthesis of Hybrid Zinc-Based Materials from Ionic Liquids: A Novel Route to Prepare Active Zn Catalysts for the Photoactivation of Water and Methane.
ACS Sustain. Chem. Eng. 2019,
7, 8090–8098.
[Google Scholar]
-
68.
Chai Y, Tang S, Wang Q, Wu Q, Liang J, Li L. Gold nanoparticles supported on ZnGa
2O
4 nanosheets as efficient photocatalysts for selective oxidation of methane to ethane under ambient conditions.
Appl. Catal. B Environ. 2023,
338, 123012.
[Google Scholar]
-
69.
Fei M, Williams B, Wang L, Li H, Yuan Y, Wilkes JR, et al. Highly Selective Photocatalytic Methane Coupling by Au-Modified Bi
2WO
6.
ACS Catal. 2024,
14, 1855–1861.
[Google Scholar]
-
70.
Zhang J, Zhang J, Shen J, Li D, Long J, Dai W, et al. Regulation of Oxygen Activation Pathways to Optimize Photocatalytic Methane Oxidative Coupling Selectivity.
ACS Catal. 2024,
14, 3855–3866.
[Google Scholar]
-
71.
Sun X, Liu G, Shen T, Hu Y, Song Z, Wu Z, et al. Directional Activation of Oxygen by the Au-Loaded ZnAl-LDH with Defect Structure for Highly Efficient Photocatalytic Oxidative Coupling of Methane. Small 2024. https://doi.org/10.1002/smll.202310857.
-
72.
Li X, Xie J, Rao H, Wang C, Tang J. Platinum- and CuO
x-Decorated TiO
2 Photocatalyst for Oxidative Coupling of Methane to C
2 Hydrocarbons in a Flow Reactor.
Angew. Chem. Int. Ed. 2020,
59, 19702–19707.
[Google Scholar]
-
73.
Yang J, Wang C, Xing J, Tang J. Palladium decorated anatase for photocatalytic partial oxidation of methane to ethane.
Surf. Interfaces 2023,
40, 103108.
[Google Scholar]
-
74.
Li X, Wang C, Yang J, Xu Y, Yang Y, Yu J, et al. PdCu nanoalloy decorated photocatalysts for efficient and selective oxidative coupling of methane in flow reactors.
Nat. Commun. 2023,
14, 6343.
[Google Scholar]
-
75.
Wang C, Li X, Ren Y, Jiao H, Wang FR, Tang J. Synergy of Ag and AgBr in a Pressurized Flow Reactor for Selective Photocatalytic Oxidative Coupling of Methane.
ACS Catal. 2023,
13, 3768–3774.
[Google Scholar]
-
76.
Song S, Song H, Li L, Wang S, Chu W, Peng K, et al. A selective Au−ZnO/TiO
2 hybrid photocatalyst for oxidative coupling of methane to ethane with dioxygen.
Nat. Catal. 2021,
4, 1032–1042.
[Google Scholar]
-
77.
Hu D, Dong C, Belhout S, Shetty S, Ng H, Brasseur P, et al. Roles of titania and plasmonic gold nanoparticles of different sizes in photocatalytic methane coupling at room temperature.
Mater. Today Energy 2023,
36, 101358.
[Google Scholar]
-
78.
Li X, Li C, Xu Y, Liu Q, Bahri M, Zhang L, et al. Efficient hole abstraction for highly selective oxidative coupling of methane by Au-sputtered TiO
2 photocatalysts.
Nat. Energy 2023,
8, 1013–1022.
[Google Scholar]
-
79.
Chen Y, Zhao Y, Liu D, Wang G, Jiang W, Liu S, et al. Continuous Flow System for Highly Efficient and Durable Photocatalytic Oxidative Coupling of Methane.
J. Am. Chem. Soc. 2024,
146, 2465–2473.
[Google Scholar]
-
80.
Deng G, Li K, Gu Z, Zhu X, Wei Y, Cheng X, et al. Synergy effects of combined red muds as oxygen carriers for chemical looping combustion of methane.
Chem. Eng. J. 2018,
341, 588–600.
[Google Scholar]
-
81.
Lambert A, Delquié C, Clémeneçon I, Comte E, Lefebvre V, Rousseau J, et al. Synthesis and characterization of bimetallic Fe/Mn oxides for chemical looping combustion.
Energy Procedia 2009,
1, 375–381.
[Google Scholar]
-
82.
Zheng Y, Li K, Wang H, Zhu X, Wei Y, Zheng M, et al. Enhanced Activity of CeO
2−ZrO
2 Solid Solutions for Chemical-Looping Reforming of Methane via Tuning the Macroporous Structure.
Energy Fuels 2016,
30, 638–647.
[Google Scholar]
-
83.
Haribal VP, Wang X, Dudek R, Paulus C, Turk B, Gupta R, et al. Modified Ceria for “Low-Temperature” CO
2 Utilization: A Chemical Looping Route to Exploit Industrial Waste Heat.
Adv. Energy Mater. 2019,
9, 1901963.
[Google Scholar]
-
84.
Wang P, Zhao G, Wang Y, Lu Y. MnTiO
3-driven low-temperature oxidative coupling of methane over TiO
2-doped Mn
2O
3-Na
2WO
4/SiO
2 catalyst.
Sci. Adv. 2017,
3, e1603180.
[Google Scholar]
-
85.
Liu Y, Qin L, Cheng Z, Goetze JW, Kong F, Fan JA, et al. Near 100% CO selectivity in nanoscaled iron-based oxygen carriers for chemical looping methane partial oxidation.
Nat. Commun. 2019,
10, 5503.
[Google Scholar]
-
86.
Li L, Cai Y-Y, Li G-D, Mu X-Y, Wang K-X, Chen J-S. Synergistic Effect on the Photoactivation of the Methane C—H Bond over Ga
3+-Modified ETS-10.
Angew. Chem. Int. Ed. 2012,
51, 4702–4706.
[Google Scholar]
-
87.
Yu X, Zholobenko VL, Moldovan S, Hu D, Wu D, Ordomsky VV, et al. Stoichiometric methane conversion to ethane using photochemical looping at ambient temperature.
Nat. Energy 2020,
5, 511–519.
[Google Scholar]
-
88.
Zheng K, Zhang X, Hu J, Xu C, Zhu J, Li J, et al. High-rate CH
4-to-C
2H
6 photoconversion enabled by Au/ZnO porous nanosheets under oxygen-free system.
Sci. China Chem. 2024,
67, 869–875.
[Google Scholar]
-
89.
Zhang H, Zhong W, Gong Q, Sun P, Fei X, Wu X, et al. Photo-Driven Iron-Induced Non-Oxidative Coupling of Methane to Ethane.
Angew. Chem. Int. Ed. 2023,
62, e202303405.
[Google Scholar]
-
90.
Jiang W, Low J, Mao K, Duan D, Chen S, Liu W, et al. Pd-Modified ZnO−Au Enabling Alkoxy Intermediates Formation and Dehydrogenation for Photocatalytic Conversion of Methane to Ethylene.
J. Am. Chem. Soc. 2021,
143, 269–278.
[Google Scholar]
-
91.
Liu Y, Chen Y, Jiang W, Kong T, Camargo PHC, Gao C, et al. Highly Efficient and Selective Photocatalytic Nonoxidative Coupling of Methane to Ethylene over Pd−Zn Synergistic Catalytic Sites.
Research 2022,
2022, 9831340.
[Google Scholar]
-
92.
Zhang W, Fu C, Low J, Duan D, Ma J, Jiang W, et al. High-performance photocatalytic nonoxidative conversion of methane to ethane and hydrogen by heteroatoms-engineered TiO
2.
Nat. Commun. 2022,
13, 2806.
[Google Scholar]
-
93.
Ma Z, Chen Y, Gao C, Xiong Y. Engineering surface lattice hydroxyl groups toward highly efficient photocatalytic methane coupling.
Chem. Commun. 2024,
60, 1132–1135.
[Google Scholar]
-
94.
Yu X, De Waele V, Löfberg A, Ordomsky V, Khodakov AY. Selective photocatalytic conversion of methane into carbon monoxide over zinc-heteropolyacid-titania nanocomposites.
Nat. Commun. 2019,
10, 700.
[Google Scholar]
-
95.
Zhang W, Xi D, Chen Y, Chen A, Jiang Y, Liu H, et al. Light-driven flow synthesis of acetic acid from methane with chemical looping.
Nat. Commun. 2023,
14, 3047.
[Google Scholar]
-
96.
Clavero C. Plasmon-induced hot-electron generation at nanoparticle/metal-oxide interfaces for photovoltaic and photocatalytic devices.
Nat. Photonics 2014,
8, 95–103.
[Google Scholar]
-
97.
Sarina S, Zhu H-Y, Xiao Q, Jaatinen E, Jia J, Huang Y, et al. Viable Photocatalysts under Solar-Spectrum Irradiation: Nonplasmonic Metal Nanoparticles.
Angew. Chem. Int. Ed. 2014,
53, 2935–2940.
[Google Scholar]
-
98.
Aslam U, Rao VG, Chavez S, Linic S. Catalytic conversion of solar to chemical energy on plasmonic metal nanostructures.
Nat. Catal. 2018,
1, 656–665.
[Google Scholar]
-
99.
Zhang Y, He S, Guo W, Hu Y, Huang J, Mulcahy JR, et al. Surface-Plasmon-Driven Hot Electron Photochemistry.
Chem. Rev. 2018,
118, 2927–2954.
[Google Scholar]